
Synthesis of novel porous graphene nanocomposite and its use as electrode and absorbent
Abstract
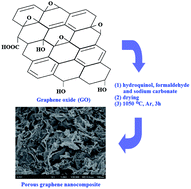
A novel three-dimensional nanostructured porous graphene nanocomposite was synthesized by sol–gel polymerization of hydroquinol (H) and formaldehyde (F) with sodium carbonate as a catalyst in graphene oxide (GO) suspension, followed by carbonization of HF and thermal reduction of GO to graphene at high temperature. The resulting material shows a BET surface area of 427 m2 g−1 and a total volume of 1.2764 cm3 g−1 with abundant mesopores. The porous graphene nanocomposite (HFG) can be used as anode for lithium ion batteries due to the porosity and thermal stability. Also, with the strong surface hydrophobicity, high mesopore ratio and pore volume, the HFG shows good absorption capacity for various organics, which makes the HFG a candidate for removal of organic contaminants from water.
 Please wait while we load your content...
Please wait while we load your content...

