Chitin hybrid materials reinforced with graphene oxide nanosheets: chemical and mechanical characterisation†
Joaquín Antonio Gonzáleza,
María Florencia Mazzobreb,
María Emilia Villanuevaa,
Luis Eduardo Díaza and
Guillermo Javier Copello*a
aCátedra de Química Analítica Instrumental, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires (UBA), IQUIMEFA (UBA-CONICET), Junín 956, C1113AAD Buenos Aires, Argentina. E-mail: gcopello@ffyb.uba.ar; Fax: +54 11 49648254; Tel: +54 11 49648254
bDepartamento de Industrias, Facultad de Ciencias Exactas y Naturales – Universidad de Buenos Aires y CONICET, Intendente Guiraldes 2160, CP 1428, Buenos Aires, Argentina
First published on 18th February 2014
Abstract
Chitin hybrid materials reinforced with graphene oxide nanosheets (nGO) have been prepared. The chitin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nGO ratio ranged from proportions where chitin was the main component to ones where nGO exceeded chitin. SEM and TEM images showed that high proportions of nGO may result in nanosheet association. FTIR, 13C solid-state NMR and DSC analyses showed that the interaction among the components would not involve the formation of new molecular bonds. nGO was shown to act as a filler that induces structural rearrangements in chitin which lead to new hydrogen bonds among the chains. The mechanical stability proved to be higher when the nGO content in the hybrid was similar to or higher than that of chitin. The rheological behaviour of the material was shown to become more solid-like with increasing nGO content. The nGO did not interfere with lysozyme activity on chitin chains, indicating that these materials would be biodegradable.
:nGO ratio ranged from proportions where chitin was the main component to ones where nGO exceeded chitin. SEM and TEM images showed that high proportions of nGO may result in nanosheet association. FTIR, 13C solid-state NMR and DSC analyses showed that the interaction among the components would not involve the formation of new molecular bonds. nGO was shown to act as a filler that induces structural rearrangements in chitin which lead to new hydrogen bonds among the chains. The mechanical stability proved to be higher when the nGO content in the hybrid was similar to or higher than that of chitin. The rheological behaviour of the material was shown to become more solid-like with increasing nGO content. The nGO did not interfere with lysozyme activity on chitin chains, indicating that these materials would be biodegradable.
Introduction
Chitin is the second most abundant biopolymer found in nature after cellulose and can be found in fungi, the exoskeleton of insects and the shells of crustaceans, including shrimp and crab, as well as other invertebrates, such as marine sponges.1,2 It is commercially available as a by-product from the food industry with little processing and, therefore, is a low-cost biopolymer.3,4 It is a polysaccharide with a structure consisting predominantly of unbranched chains of β-(1→4)-2-acetoamido-2-deoxy-D-glucose. In the past decade it has sparked interest for use as a building block for materials to be applied in a wide range of fields such as medicine, biotechnology, biosorption and the food industry.5 This interest comes from its biocompatibility, biodegradability, low toxicity and capacity to form mechanically stable materials that can be used for protein sorption, water remediation, food wrapping, tissue engineering, wound dressing and drug delivery systems.3,6–9 Although it is still a developing field, chitin-based materials reports have been increasing since researchers have understood chitin's chemical behaviour and developed promising techniques to obtain these materials. Among these methods, the isolation of 3D chitin scaffolds from marine sponges and the biomimetic techniques derived from this, the formation of chitin hydrogels for biomedical purposes, the formation of cross-linked chitin hydrogels for dye sorption and the dissolution of chitin in ionic liquids for electrospinning should all be mentioned.1,6,10–13Polysaccharides and other natural polymers are usually chemically modified in order to tailor their physical and chemical properties, such as mechanical strength, thermal stability or chemical reactivity.14 The development of hybrid materials by the incorporation of fillers has been documented as a promising strategy for obtaining improved materials.15–18 Some examples are the use of magnetite as nanofiller for carrageenans,19 cellulose whiskers for alginate-acerola puree,20 and carbon nanotubes and clay for chitosan.21,22
Among these approaches, special attention has been paid to the use of graphene oxide (GO) in polymer reinforcement and chemical modification. GO can be obtained from graphite by assorted methods, among which Hummer's oxidative exfoliation is the most widespread technique. Graphite consists of stacked nano-sheets of graphene, a single layered 2D hexagonal arrangement of sp2 hybridized carbon atoms.23,24 It is considered a good alternative to the use of its allotropes, the carbon nanotubes, not only because it can be cheaply obtained from low-cost graphite but also because it has shown higher biocompatibility.25 Pinto et al. reviewed the biocompatibility of graphene based materials and concluded that the introduction of hydrophilic groups needs to be considered when trying to obtain biocompatible materials.26 In addition, unlike graphene, GO has in its structure a large number of functional groups such as hydroxyls, epoxides, carbonyls and carboxyls, which endow it with chemical reactivity.27 Chemically exfoliated GO can be obtained with controlled degrees of oxidation by the Modified Hummers' method.28 Also, reduced GO can be obtained by tailored chemical and thermal reduction.29 These characteristics make GO a promising biopolymer filler for a wide range of applications.30,31 Several researchers have reported the use of GO as a filler for polysaccharide based materials, such as starch, alginate, agarose, cellulose and chitosan, mainly aiming for the reinforcement of mechanical properties. There are also studies suggesting the improvement of metal sorption capacity, electrical conductivity and enzyme immobilization performance.32–41
This study was motivated by the potential applications of combined chitin–GO nanosheets. In addition to mechanical reinforcement, the resulting material would have straightforward applications in the biomedical field and food industry, such as drug delivery and wound dressing, since both are biocompatible and non-toxic.3,6 Chitin itself has been used in the food industry for several applications including the formation of biodegradable films, recovery of waste material from food processing discards, immobilisation of enzymes and recovery of proteins for processing purposes.4,7 On the other hand, graphene oxide, less studied for its applications in the food industry than chitin, has promising properties for the development of food wraps for example, due to its oxygen barrier behaviour.42 In addition, the chemical groups in GO sheets allow for joint applications with chitin, a polysaccharide which is not very reactive. Recently, Gou et al. reported that GO enhances the mechanical properties of chitin films obtained by the dissolution of the polysaccharide with NaOH/urea.43 Our work shows the properties of a gel-like hybrid material made of chitin, dissolved using a methanol–calcium solvent, and reinforced with GO nanosheets. Herein we present the influence of the chitin:GO ratio on the chemical, structural and mechanical properties of the final material. Chitin:GO ratios ranged from higher levels of chitin than GO to ones where the GO levels were higher. The hybrids were characterized by Fourier Transform Infrared Spectroscopy, Differential Scanning Calorimetry, Scanning and Transmission Electron Microscopy, 13C solid-state Nuclear Magnetic Resonance and rheological measurements. The water content, water uptake and biodegradability were also studied.
Experimental section
Materials
Natural graphite powder (<125 μm particle size) was purchase from Bitter (UK). Chitin from crab shells (DA: 92%; Mr ≈ 400000) was obtained from Fluka (USA). Calcium chloride dihydrate and methanol were purchased from J.T. Baker (N.J., USA). Water was filtered and deionized with a Milli-Q, Millipore system (Milford, MA, USA). All other reagents were of analytical grade.
Preparation of graphene oxide nanosheets
GO nanosheets were prepared through Hummers' method.44 In the current synthesis, graphite powder (2.0 g) and sodium nitrate (1.0 g) were mixed with sulfuric acid (46 mL) in an ice bath with sustained agitation for 4 h. Then, potassium permanganate (6.0 g) was added under stirring. The reaction mixture was kept at 35 °C for 2 h. Afterwards, 92 mL of water was added, keeping the solution in the ice bath, and the mixture was further diluted by the addition of 200 mL of water after keeping the temperature at 98 °C for another 2 h. Hydrogen peroxide (20 mL) was then added to reduce the residual potassium permanganate and stirring continued until the mixture turned brown. Finally, the mixture was centrifuged to obtain the graphite oxide powder. In order to clean out any remnants of salt and acid, the powder was re-dispersed in ultra-pure water and re-centrifuged several times. The powder was finally dried at 80 °C.The graphite oxide was exfoliated into GO monolayer nanosheets (nGO) by sonication at 35 kHz for 30 min after dispersion in citrate buffer (0.4 M; pH: 4.2).45 Then, the suspension was centrifuged and the pellet was washed with water and then with methanol. The methanol was removed by heating in a stove at 60 °C and the graphene oxide powder was then stored at room temperature.
Each batch of synthesized GO nanosheets was analysed by Dynamic Light Scattering (DLS, Zetasizer Nano-Zs, Malvern Instruments, Worcestershire, UK). The GO nanosheets hydrodynamic diameter was found to be between 370 and 400 nm in all syntheses. The yield of the oxidized graphene can be judged by its carbon-to-oxygen ratio.44 The carbon-to-oxygen ratio of the sample was 1.7.
Preparation of chitin hydrogel and chitin–nGO hybrid materials
To prepare a transparent calcium solvent, 42.5 g of calcium chloride dihydrate was suspended in 50 mL of methanol and refluxed for 30 min at 82 °C to a state of near-dissolution. One gram of chitin powder was suspended in the calcium solvent and refluxed for 2 h at 90 °C with stirring.46Different mass ratios of chitin and nGO were mixed by thorough agitation in order to obtain six types of hybrid materials with different chitin to graphene oxide ratios (Chi:nGO): (24:1), (12:1), (3:1), (1.2:1), (0.6:1) and (0.3:1). At the higher ratios Chi is the major component and in the lower ratios it is the lesser. Since various properties and applications could arise from both components, by studying this range of compositions it was expected that a full coverage of the effect of nGO on Chi gels could be achieved.
The chitin–nGO mixtures were poured between two glasses spaced by glass slides of known width and then submerged in methanol until they gelled. Finally, the gels were subjected to several water incubations in order to wash out all of the methanol and CaCl2 residues. Blank chitin gels without nGO were obtained by a similar procedure and named Chi.
Characterisation of the hybrid material
| Wu (%) = (W1 − W0)/W0 × 100% | (1) |
Amplitude sweeps were performed first in order to determine the linear viscoelastic range (LVR). Storage (G′) and loss (G′′) shear moduli as well as strain were recorded as a function of stress, at a constant frequency of 1 Hz and temperature of 20 °C. The constant strain value, at which the following frequency sweeps were performed to obtain the mechanical spectra, was chosen from the originally determined LVR for each sample. Each mechanical spectrum was then recorded at this constant strain value (γ = 0.05%) in the LVR: G′ and G′′, and the tangent of the phase angle (tanδ = G′′/G′) and complex viscosity (η*) were obtained as a function of increasing angular frequency (ω; rad s−1), after reaching steady-state conditions for each point.
Results and discussion
Macro and microscopical characterisation
The material obtained is a gel thick enough to withstand handling. ESI 1† shows that the macroscopical stiffness is visually higher in those gels with a higher nGO content. SEM images were used to observe the topography of the materials and how it is altered by the addition of nGO (Fig. 1). Chitin has a smooth and homogeneous surface that is lost as the amount of the inorganic compound increases. ESI 2† shows TEM images of Chi and Chi–nGO hybrids. The chitin gel shows a homogeneous network, while those with increasing amounts of nGO show more dense areas with the loss of homogeneity. These dark spots are attributed to accumulations of graphene oxide. In the image of the (24:1) hybrid one could observe isolated nanosheets, which become more difficult to find at higher percentages; this observation leads to the assumption that the GO is homogeneously dispersed when it is used at a low proportion. Therefore, both types of microscopy demonstrate that when the amount of graphene oxide is increased it is difficult to achieve a good dispersion of the nanosheets and they tend to self-associate, forming dark accumulations visible in TEM images. Other researchers have proposed that GO acts as a filler in chitosan–GO composites which could also be the case for these Chi–nGO hybrids.15 Guo et al. observed a similar behaviour when the nGO content was increased in chitin films and also propose that nGO acts as a filler in chitin–nGO films.43
 | ||
| Fig. 1 SEM images of the Chi and Chi–nGO hybrids. | ||
Water content and water uptake
Table 1 presents the water content and water uptake (Wu) results for the different hybrid compositions. The variation in water content was statistically analysed by one way ANOVA followed by a Tukey post-test. The water content determination shows that when the amount of nGO increases, the water content of the material decreases significantly when a Chi hydrogel is compared with Chi–nGO hybrids (3:1) (p < 0.05) and from (1.2:1) to lower ratios (p < 0.01). Also, between Chi:nGO (3:1) and (0.3:1) there is a significant drop in the water content (p < 0.01), as well as between (0.6:1) and (0.3:1) (p < 0.05). This probably occurs because the nGO acts as a large nanofiller in the chitin matrix and replaces the pore free water without any swelling effect. On the other hand, although the Wu percentage is inversely proportional to the amount of nGO, the drop in Wu is not steep enough to present significant differences among the hybrids (p > 0.05, non-parametric Kruskal–Wallis test). This could be explained in terms of two events; first, the large size of the GO nanosheets (370–400 nm) prevents the entry of water molecules during re-hydration; and second, after the material dries its size decreases considerably, so this could be accompanied by a rearrangement of the chitin chains around the nGO that hinders the hydration process.
Spectroscopic characterisation
The FTIR spectra of graphite and graphene oxide are shown in ESI 3.† The graphene oxide spectrum shows an increase in the bands corresponding to oxidized groups, which confirms the chemical exfoliation of graphite. In the spectrum of nGO, the band at 1240 cm−1 is attributed to the C–O–C bond stretching which demonstrates the formation of epoxy groups. The presence of carboxyl and carbonyl functional groups can also be detected at 1400 cm−1 and 1720 cm−1, which correspond to C–OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, respectively.33 Fig. 2 shows the spectra of nGO, Chi and Chi–nGO hybrids. The Chi spectrum shows a band at 1660–1620 cm−1 corresponding to the amide I band; this band involves a doublet at 1655 and 1625 cm−1 (C–O and C–N stretching, respectively). Other characteristic bands are the amide II band (1540 cm−1, N–H stretching) that is detectable together with the amide III band (1390 cm−1) and the glycosidic bond band at 900–1100 cm−1 (C–O–C stretching).48 Comparing the spectra, one can see the increase and decrease of the relative intensities of the characteristic bands of each component according to their proportion in the material. For example, as nGO increases the intensity of its characteristic bands also increases. In the spectra of the hybrids one could not observe any new bands or the disappearance of any bands present in the individual spectra of Chi or nGO. This indicates that the interaction between chitin and nGO probably does not involve the formation of a new functional group detectable by the FTIR experimental conditions. The FTIR spectra of the other Chi–nGO hybrids are shown in ESI 4.†
O stretching, respectively.33 Fig. 2 shows the spectra of nGO, Chi and Chi–nGO hybrids. The Chi spectrum shows a band at 1660–1620 cm−1 corresponding to the amide I band; this band involves a doublet at 1655 and 1625 cm−1 (C–O and C–N stretching, respectively). Other characteristic bands are the amide II band (1540 cm−1, N–H stretching) that is detectable together with the amide III band (1390 cm−1) and the glycosidic bond band at 900–1100 cm−1 (C–O–C stretching).48 Comparing the spectra, one can see the increase and decrease of the relative intensities of the characteristic bands of each component according to their proportion in the material. For example, as nGO increases the intensity of its characteristic bands also increases. In the spectra of the hybrids one could not observe any new bands or the disappearance of any bands present in the individual spectra of Chi or nGO. This indicates that the interaction between chitin and nGO probably does not involve the formation of a new functional group detectable by the FTIR experimental conditions. The FTIR spectra of the other Chi–nGO hybrids are shown in ESI 4.†
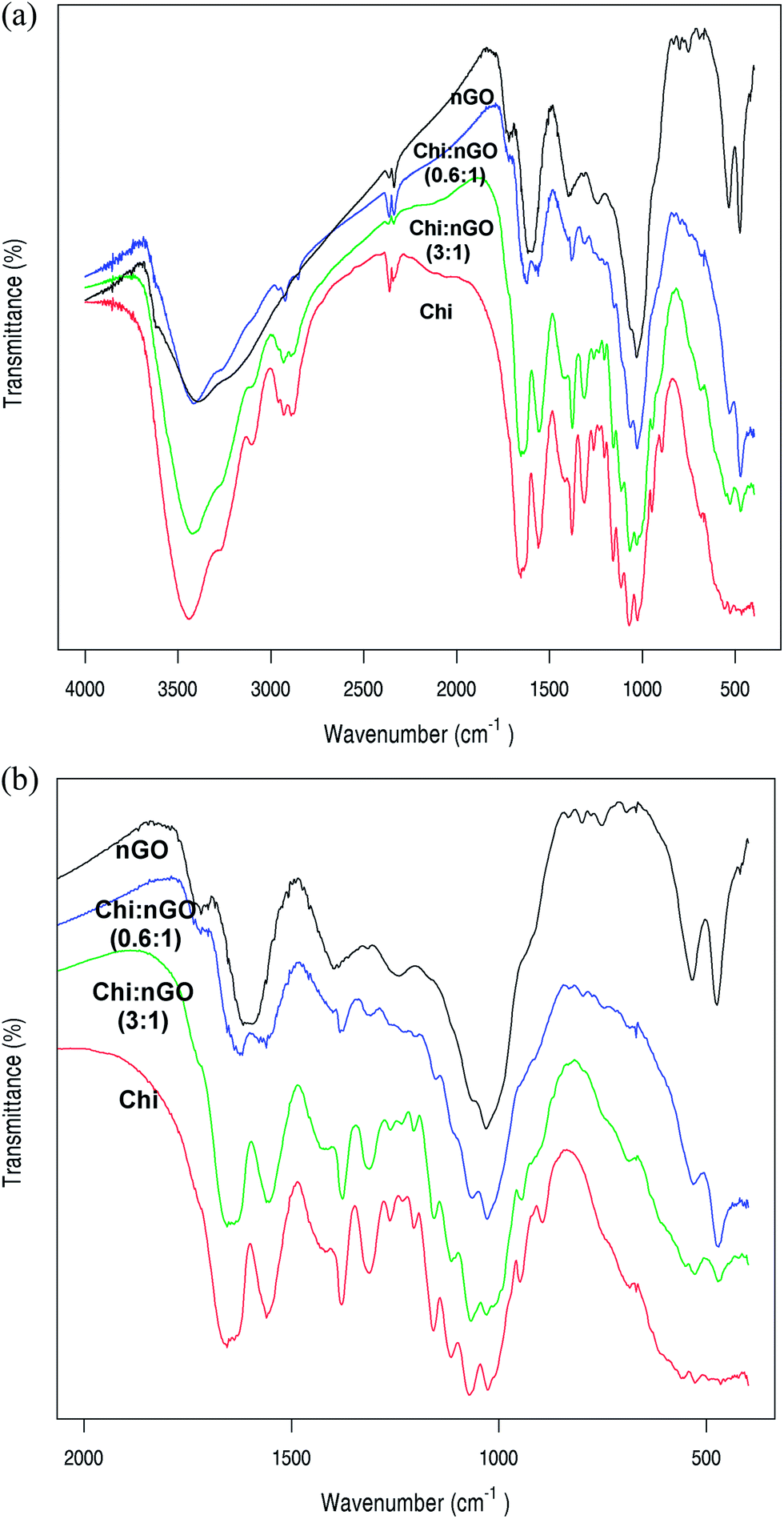 | ||
| Fig. 2 (a) FTIR spectra of nGO, Chi and Chi–nGO hybrids from 400 to 4000 cm−1. (b) FTIR spectra of nGO, Chi and Chi–nGO hybrids from 400 to 2000 cm−1. | ||
The 13C CP-MAS NMR spectra of commercial chitin, Chi and Chi–nGO hybrids are shown in Fig. 3. As in the FTIR spectra, no additional signals or peak shifts can be observed. On the other hand, the relative intensity of the 73 and 76 ppm signals, corresponding to C3 and C5, varies among the spectra. Crab shell chitin, used in this study, appears in the form of α-chitin for which the typical solid NMR spectrum shows a higher signal for C3 than for C5 in a well-defined doublet.49,50 This is also observed in the spectrum of commercial chitin powder. However, the spectrum of the Chi hybrid shows similar intensities for the C3 and C5 signals. A similar intensity of these signals could also be seen in the spectra of the Chi–nGO hybrids. Also, along with the increase in nGO content, the doublet tends to merge into one signal due to peak broadening. The literature reports that when comparing the 13C CP-MAS NMR spectra of α, β and γ-chitin, the main differences appear in the C3 and C5 signals when their relative intensities are compared.49,50 The variation of these signals was attributed to the possibility of the formation of different intra and inter-chain hydrogen bonds in the three polymorphic crystalline structures of chitin. Therefore, comparing the FTIR and NMR spectra, it could be proposed that in the process of suspending the chitin, the addition of nGO and the subsequent gelling, the α-chitin structure is lost and a structural rearrangement with new hydrogen bonds takes place. In fact, as can be seen in ESI 5,† the simple presence of nGO is not the reason for the change in C3 and C5 signals since the spectrum of a mixture of chitin and nGO powders shows no difference from the commercial chitin powder spectrum. Moreover, the C3 and C5 signal broadening observed in the spectra of Chi–nGO hybrids is not seen in the spectrum of the mixed powders, which demonstrates the influence of the nGO on the hybrids’ structure. Hence, after the loss of the α-chitin structure, the nGO promoted rearrangement of chitin chains is probably the reason for the reinforcement of the hybrids since no chemical bonds or interactions could be demonstrated by either FTIR or solid NMR spectroscopy. On the other hand, considering the presence of hydrophilic groups in both components, the possibility of the formation of hydrogen bonds between chitin and nGO cannot be neglected. As has been pointed out by other researchers working on chitin based materials, the lack of any visible changes in the measured spectra indicates that there are no strong interactions among the components but weak interactions, such as hydrogen bonds, could be expected.10,51
 | ||
| Fig. 3 Solid-state 13C-NMR spectra of Chi, Chi–nGO hybrid and commercial chitin powder. | ||
Regarding nGO, it would be expected that if both components of the hybrid were present in similar proportions, the signals from nGO would appear in the spectra of the Chi–nGO hybrids. These signals are not visible in any of the spectra. Also, the acquisition of a pure nGO powder spectrum with the same number of scans as the hybrids showed no clear peaks (data not shown). In solid state NMR, the homotopic carbons of large and flat molecules, such as nGO, are more likely to have different effective magnetic fields (depending on the carbon position in the molecule), resulting in differences in their chemical shifts.52 These would lead to broad bands and lower sensitivity for nGO than for chitin.
Thermal transitions by Differential Scanning Calorimetry
Fig. 4 shows the DSC thermograms obtained for Chi, Chi–nGO hybrid materials and the commercial chitin powder. The Chi thermogram shows a broad endothermic peak around 60 °C and an exothermic event at a temperature higher than 300 °C. The endothermic peak was attributed by other authors to the evaporation of water bound to the polar sites of chitin, changes in this transition were associated with different polymorphic crystalline structures (α, β and γ) of this polysaccharide.50 This transition was also observed in the thermograms of both the Chi hydrogel and the commercial chitin powder at a similar temperature. The endothermic peak was also observed in the hybrid materials, with a shift towards higher temperatures as the nGO content in the system increases. For the Chi:nGO (0.3:1) hybrid the endothermic peak occurs at the highest temperature (96 °C), suggesting structural changes in the hybrid material that increase water–solid interactions. This is consistent with the solid state NMR spectra results and supports the premise that the ratio of nGO is involved in the structural arrangement of chitin chains in the hybrids. Near 200 °C a sharp exothermic event was observed in the nGO-containing systems. This peak corresponds to the decomposition of the oxygen groups of the nGO.53 As expected, the area of the peak increases with the amount of nGO in the hybrids and was not observed for the chitin hydrogel.
 | ||
| Fig. 4 DSC thermograms of Chi and Chi–nGO hybrids. | ||
The oxidation of the samples was observed as a sharp increase in the energy flow due to the exothermic nature of the oxidation reactions. The onset temperature of the oxidative reaction (OOT) was taken as the intersection of the extrapolated baseline and the tangent line (leading edge) of the exothermal peak. Fig. 4 shows that the oxidation process was only evident in those samples that did not undergo decomposition (Chi: absence of exothermic peak at 200 °C) or were partially decomposed during heating (Chi:nGO (12:1) and (1.2:1)). In the commercial chitin thermogram this reaction is observed as a rise of the baseline after a sharp exothermic event around 340 °C, which other researchers have attributed to the decomposition of the acetyl residues of chitin.54 This peak is absent in the thermograms of Chi and the Chi–nGO hybrids. This decomposition probably does not occur as a sharp and defined event due to the structural arrangement of the chitin chains and is observed as a part of the exothermic increase of energy flow mentioned above.
DSC thermograms were also carried under a protective N2 atmosphere in order to study phase transitions of the materials (ESI 6†). In the chitin powder thermogram a sharp endothermic event (Tm) could be observed at 370 °C. For Chi and Chi:nGO hybrids this event was not evident. For Chi thermograms this peak is observed at 310 °C. Again, probably this change in the transition temperature could be due to a new chitin structural arrangement.50
Mechanical stability
Fig. 5 shows the remaining weight of the hybrids after incubation with mechanical stirring. An important weight loss is observed in the Chi, and Chi:nGO (24:1) and (12:1) hybrids after 24 h due to a random rupture of the composites. The weight losses of the hybrids with higher nGO contents were lower, which was probably due to the detachment of small peripheral fragments rather than rupture of the hybrids. The weight loss for ratios from (3:1) to (0.3:1) were not significantly different (p > 0.05, one way ANOVA). Also, the standard deviation of the weight loss of these hybrids proved to be lower, indicating they are more resistant materials. Therefore, it can be concluded that the cohesion of the material and its mechanical stirring stress resistance is proportional to the amount of nGO up to a Chi:nGO ratio of (12:1), with no significant improvement with higher nGO contents.
 | ||
| Fig. 5 Remaining weight of the hybrids after incubation with mechanical stirring. | ||
Rheological behaviour
For all composites the G′ behaviour was independent of frequency in the tested range (Fig. 6). Also G′ was greater than the G′′ for all cases, which implies that the elastic component of the material is dominant over the viscous component, which is typical of a gel-like behaviour. Table 2 shows a comparison of the rheological parameters measured at 10 Hz and γ = 0.05%. All of the hybrids show similar values of G′ without a trend associated with the nGO content. On the other hand, the tanδ values decrease up to 25% for the higher nGO contents. For gel materials with values of tanδ below 1 this drop is indicative of a more solid-like behaviour.55 The complex viscosity of the hybrids decreases almost linearly with the increase of the frequency, showing shear thinning behaviour which is probably due to the structure of the hybrid polymer network (ESI 7†).
| Parameter | Hybrid | ||||||
|---|---|---|---|---|---|---|---|
| Chi | Chi–nGO | ||||||
| 24:1 |
12:1 |
3:1 |
1.2:1 |
0.6:1 |
0.3:1 |
||
| G′ (kPa) | 26.2 ± 0.9 | 19 ± 2 | 20 ± 2 | 14 ± 2 | 23.4 ± 0.9 | 45 ± 9 | 27.1 ± 0.7 |
| G′′ (kPa) | 4.1 ± 0.3 | 2.9 ± 0.4 | 3.0 ± 0.3 | 1.9 ± 0.3 | 3.4 ± 0.2 | 5 ± 1 | 3.3 ± 0.2 |
| Tanδ |
0.156 ± 0.004 | 0.151 ± 0.007 | 0.151 ± 0.003 | 0.1365 ± 0.0007 | 0.144 ± 0.005 | 0.115 ± 0.001 | 0.12 ± 0.01 |
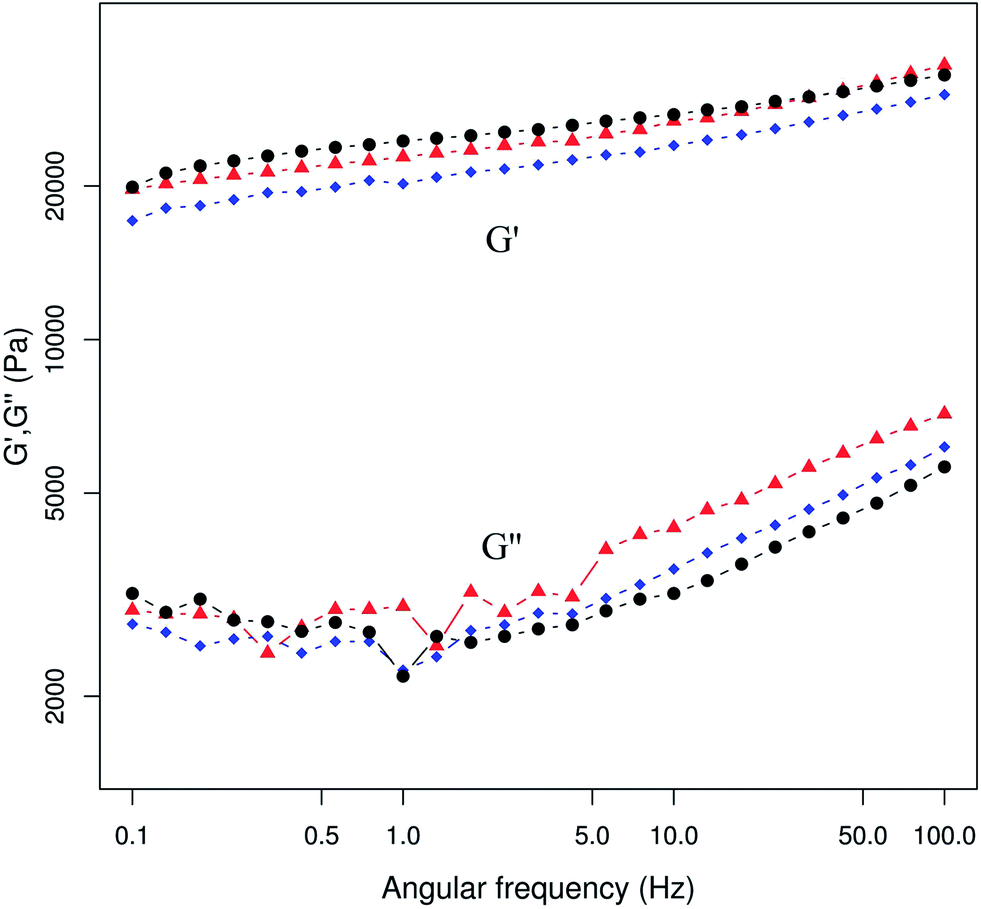 | ||
| Fig. 6 Storage modulus (G′) and loss modulus (G′′) frequency dependence of the hybrids. Symbols: Chi (▲), Chi:nGO (1.2:1) (♦), and Chi:nGO (0.3:1) (●). | ||
In vitro biodegradability
It is well known that chitin is a biodegradable polysaccharide with a glycosidic bond that could be hydrolysed by several enzymes present in the environment, i.e. lysozyme and chitinase.47 ESI 8† shows the hybrid discs before and after lysozyme exposure. After exposure to lysozyme, it is possible to see significant rupturing of all Chi–nGO discs regardless of the proportion of each component. This suggests that the nGO content does not affect or interfere with the activity of lysozyme on chitin. In fact, the degree of structural rupturing of the hybrids did not allow weighing of the discs as they disaggregated upon contact. This also indicates that the nGO is acting as a nanofiller rather than interacting strongly with the chitin chains.Conclusions
In this study, hybrid materials made of chitin reinforced with GO nanosheets have been successfully prepared. The structures and mechanical properties of the hybrid materials proved to be dependent on the chitin to nGO ratio. These hybrid nanocomposites showed that the structural network of chitin can be loaded with nGO masses over three times the amount possible with the polysaccharide. Microscopy images showed that high proportions of nGO may result in the association of nanosheets. Spectroscopic and DSC analyses showed that the interaction among the components did not involve the formation of new molecular bonds. On the other hand, nGO would act as a filler that induces structural rearrangements in chitin chains, causing new hydrogen bonds between the chains which lead to changes in the mechanical properties of the materials. Mechanical stability and rheological behaviour tests showed that hybrids with higher nGO contents were more stable and solid-like materials. In addition, nGO did not interfere with lysozyme activity on chitin chains, which indicates that the polymeric matrix of these materials maintains its biodegradability.Acknowledgements
J.A.G. is grateful for his doctoral fellowship granted by Universidad de Buenos Aires. M.E.V. is grateful for her doctoral fellowship granted by Consejo Nacional de Investigaciones Científicas y Tecnológicas. This work was supported with grants from Universidad de Buenos Aires (UBACYT 20020100100919 and 20020090300085). The authors would like to thank J. Nesterzak for his technical assistance and N. Dobin-Berstein for language corrections.References
- M. Wysokowski, V. V. Bazhenov, M. V. Tsurkan, R. Galli, A. L. Stelling, H. Stöcker, S. Kaiser, E. Niederschlag, G. Gärtner, T. Behm, M. Ilan, A. Y. Petrenko, T. Jesionowski and H. Ehrlich, Int. J. Biol. Macromol., 2013, 62, 94–100 CrossRef CAS PubMed.
- H. Ehrlich, M. Maldonado, K. Spindler, C. Eckert, T. Hanke, R. Born, C. Goebel, P. Simon, S. Heinemann and H. Worch, J. Exp. Zool., Part B, 2007, 308B, 347–356 CrossRef CAS PubMed.
- M. N. Ravi Kumar, React. Funct. Polym., 2000, 46, 1–27 CrossRef.
- F. Shahidi, J. K. V. Arachchi and Y.-J. Jeon, Trends Food Sci. Technol., 1999, 10, 37–51 CAS.
- R. Muzzarelli, Carbohydr. Polym., 1983, 3, 53–75 CrossRef CAS.
- H. Tamura, T. Furuike, S. V. Nair and R. Jayakumar, Carbohydr. Polym., 2011, 84, 820–824 CrossRef CAS PubMed.
- F. Wolman, G. Copello, A. Mebert, A. Targovnik, M. Miranda, A. Navarro del Cañizo, L. Díaz and O. Cascone, Eur. Food Res. Technol., 2010, 231, 181–188 CrossRef CAS PubMed.
- G. J. Copello, A. M. Mebert, M. Raineri, M. P. Pesenti and L. E. Diaz, J. Hazard. Mater., 2011, 186, 932–939 CAS.
- M. Sharma, C. Mukesh, D. Mondal and K. Prasad, RSC Adv., 2013, 3, 18149–18155 CAS.
- K. Spinde, M. Kammer, K. Freyer, H. Ehrlich, J. N. Vournakis and E. Brunner, Chem. Mater., 2011, 23, 2973–2978 CrossRef CAS.
- H. Ehrlich, P. Simon, M. Motylenko, M. Wysokowski, V. V. Bazhenov, R. Galli, A. L. Stelling, D. Stawski, M. Ilan, H. Stocker, B. Abendroth, R. Born, T. Jesionowski, K. J. Kurzydlowski and D. C. Meyer, J. Mater. Chem. B, 2013, 1, 5092–5099 RSC.
- M. Wysokowski, M. Motylenko, H. Stocker, V. V. Bazhenov, E. Langer, A. Dobrowolska, K. Czaczyk, R. Galli, A. L. Stelling, T. Behm, L. Klapiszewski, D. Ambrozewicz, M. Nowacka, S. L. Molodtsov, B. Abendroth, D. C. Meyer, K. J. Kurzydlowski, T. Jesionowski and H. Ehrlich, J. Mater. Chem. B, 2013, 1, 6469–6476 RSC.
- P. S. Barber, C. S. Griggs, J. R. Bonner and R. D. Rogers, Green Chem., 2013, 15, 601–607 RSC.
- J. Desbrieres and V. Babak, Soft Matter, 2010, 6, 2358–2363 RSC.
- Y. Pan, T. Wu, H. Bao and L. Li, Carbohydr. Polym., 2011, 83, 1908–1915 CAS.
- M. N. Anglès and A. Dufresne, Macromolecules, 2000, 33, 8344–8353 CrossRef.
- N. Lin and A. Dufresne, Macromolecules, 2013, 46, 5570–5583 CrossRef CAS.
- H. Feng, Y. Li and J. Li, RSC Adv., 2012, 2, 6988–6993 RSC.
- A. L. Daniel-da-Silva, J. Moreira, R. Neto, A. C. Estrada, A. M. Gil and T. Trindade, Carbohydr. Polym., 2012, 87, 328–335 CrossRef CAS PubMed.
- H. M. C. Azeredo, K. W. E. Miranda, H. L. Ribeiro, M. F. Rosa and D. M. Nascimento, J. Food Eng., 2012, 113, 505–510 CrossRef CAS PubMed.
- C. Tang, L. Xiang, J. Su, K. Wang, C. Yang, Q. Zhang and Q. Fu, J. Phys. Chem. B, 2008, 112, 3876–3881 CrossRef CAS PubMed.
- S.-F. Wang, L. Shen, W.-D. Zhang and Y.-J. Tong, Biomacromolecules, 2005, 6, 3067–3072 CrossRef CAS PubMed.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CAS.
- A. Y. W. Sham and S. M. Notley, Soft Matter, 2013, 9, 6645–6653 RSC.
- K.-H. Liao, Y.-S. Lin, C. W. Macosko and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CAS.
- A. M. Pinto, I. C. Gonçalves and F. D. Magalhães, Colloids Surf., B, 2013, 111, 188–202 CrossRef CAS PubMed.
- X. Yang, C. Chen, J. Li, G. Zhao, X. Ren and X. Wang, RSC Adv., 2012, 2, 8821–8826 RSC.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S.-J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS PubMed.
- C. Botas, P. Alvarez, C. Blanco, M. D. Gutierrez, P. Ares, R. Zamani, J. Arbiol, J. R. Morante and R. Menendez, RSC Adv., 2012, 2, 9643–9650 RSC.
- C. Wan, M. Frydrych and B. Chen, Soft Matter, 2011, 7, 6159–6166 CAS.
- A. He, B. Lei, C. (Sage) Cheng, S. Li, L. Ma, S. Sun and C. Zhao, RSC Adv., 2013, 3, 22120–22129 RSC.
- Y. He, N. Zhang, Q. Gong, H. Qiu, W. Wang, Y. Liu and J. Gao, Carbohydr. Polym., 2012, 88, 1100–1108 CrossRef CAS PubMed.
- R. Li, C. Liu and J. Ma, Carbohydr. Polym., 2011, 84, 631–637 CrossRef CAS PubMed.
- N. Zhang, H. Qiu, Y. Si, W. Wang and J. Gao, Carbon, 2011, 49, 827–837 CrossRef CAS PubMed.
- J.-D. Qiu, J. Huang and R.-P. Liang, Sens. Actuators, B, 2011, 160, 287–294 CrossRef CAS PubMed.
- A. A. Alhwaige, T. Agag, H. Ishida and S. Qutubuddin, RSC Adv., 2013, 3, 16011–16020 RSC.
- C.-J. Cai, M.-W. Xu, S.-J. Bao, C. Lei and D.-Z. Jia, RSC Adv., 2012, 2, 8172–8178 RSC.
- Y. Wang, P. Zhang, C. F. Liu and C. Z. Huang, RSC Adv., 2013, 3, 9240–9246 RSC.
- Y. Feng, X. Zhang, Y. Shen, K. Yoshino and W. Feng, Carbohydr. Polym., 2012, 87, 644–649 CrossRef CAS PubMed.
- D. Han, L. Yan, W. Chen, W. Li and P. R. Bangal, Carbohydr. Polym., 2011, 83, 966–972 CrossRef CAS PubMed.
- D. Han, L. Yan, W. Chen and W. Li, Carbohydr. Polym., 2011, 83, 653–658 CrossRef CAS PubMed.
- H. M. Kim, J. K. Lee and H. S. Lee, Thin Solid Films, 2011, 519, 7766–7771 CrossRef CAS PubMed.
- Y. Guo, B. Duan, J. Zhou and P. Zhu, Cellulose, 2014, 1–11 Search PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- Q. Bao, D. Zhang and P. Qi, J. Colloid Interface Sci., 2011, 360, 463–470 CrossRef CAS PubMed.
- H. Tamura, H. Nagahama and S. Tokura, Cellulose, 2006, 13, 357–364 CrossRef CAS.
- Y. M. Yang, W. Hu, X. D. Wang and X. S. Gu, J. Mater. Sci.: Mater. Med., 2007, 18, 2117–2121 CrossRef CAS PubMed.
- Y. Saito, J.-L. Putaux, T. Okano, F. Gaill and H. Chanzy, Macromolecules, 1997, 30, 3867–3873 CrossRef CAS.
- S. F. Tanner, H. Chanzy, M. Vincendon, J. C. Roux and F. Gaill, Macromolecules, 1990, 23, 3576–3583 CrossRef CAS.
- M.-K. Jang, B.-G. Kong, Y.-I. Jeong, C. H. Lee and J.-W. Nah, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3423–3432 CrossRef CAS PubMed.
- D. Schleuter, A. Günther, S. Paasch, H. Ehrlich, Z. Kljajić, T. Hanke, G. Bernhard and E. Brunner, Carbohydr. Polym., 2013, 92, 712–718 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- L. S. Guinesi and É. T. G. Cavalheiro, Thermochim. Acta, 2006, 444, 128–133 CrossRef CAS PubMed.
- M. V. Tzoumaki, T. Moschakis and C. G. Biliaderis, Carbohydr. Polym., 2013, 95, 324–331 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47986b |
| This journal is © The Royal Society of Chemistry 2014 |