A high-performance electrocatalytic air cathode derived from aniline and iron for use in microbial fuel cells
Xinhua Tangab,
Haoran Lic,
Weida Wangd,
Zhuwei Duc and
How Yong Ng*a
aCentre for Water Research, Department of Civil and Environmental Engineering National University of Singapore, Singapore 117576, Singapore. E-mail: howyongng@nus.edu.sg; Fax: +65 67744202; Tel: +65 65164777
bNUS Graduate School for Integrative Sciences and Engineering, National University of Singapore, Singapore 117456, Singapore
cNational Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China
dCivil and Environment Engineering School, University of Science and Technology Beijing, Beijing 100083, China
First published on 21st January 2014
Abstract
In this study, a high-performance electrocatalyst derived from aniline and iron was synthesized. The catalyst was prepared by pyrolyzing iron-impregnated polyaniline in an NH3 atmosphere at high temperature. Pyrolyzing in NH3 significantly increases the micropores, and the pyridinic nitrogen on the surface of the catalyst, leading to the excellent performance towards oxygen reduction. The electrocatalytic activity of this catalyst towards the oxygen reduction reaction was evaluated using a rotating ring disk electrode (RRDE). This catalyst exhibits a high onset potential of about 0.92 V (vs. reversible hydrogen electrode, RHE) and the half-wave potential difference between this catalyst and the benchmark Pt/C catalyst is only about 70 mV. The RRDE study also demonstrated that this catalyst for the four-electron reduction of oxygen has a very high selectivity. Applied as air cathodes in microbial fuel cells, the catalyst exhibits an excellent catalytic activity. The MFC with this catalyst achieves a maximum power density of 12.56 W m−3, higher than the 9.92 W m−3 of the MFC with a Pt/C catalyst and the 1.34 W m−3 of the MFC with bare graphite felt. These results demonstrate that this catalyst is an excellent alternative to the prohibitive Pt/C catalyst for practical applications.
Introduction
A microbial fuel cell (MFC) is a bioelectrochemical system that converts chemical energy in organic matter into electrical energy by the catalysis of microorganisms.1–3 As a novel biotechnology for wastewater treatment and renewable energy production at the same time, MFCs have drawn much attention in the past decade.4–6 Great progress has been made in MFCs and this technology has great potential for wastewater treatment. However, the prohibitive cost of the cathode is a pressing issue that needs to be solved. Currently, a platinum cathode is the most popular catalyst used to efficiently drive the oxygen reduction reaction in MFCs; however, it is estimated that this costly cathode accounts for up to 47% of the overall cost of an MFC system, which hinders its practical applications significantly.7 As a consequence, there is an urgent need to explore alternatives to bring down the cost.The development of a non-precious catalyst with a high catalytic activity towards oxygen reduction has recently attracted much attention not only in MFCs, but also in fuel cells.8–11 Among these reported alternatives, carbon–nitrogen–metal catalysts synthesized by pyrolyzing precursors comprising carbon, nitrogen and transition metals (Fe, Co, Ni) are considered to be the most promising candidates, because these non-precious catalysts possess excellent catalytic activity and remarkable stability for oxygen reduction reactions.8,9,12
In this study, a non-precious carbon–nitrogen–metal catalyst (P–Fe) was prepared by pyrolyzing iron incorporated polyaniline under an NH3 atmosphere. This catalyst was comprehensively investigated using a field emission scanning electron microscope (FESEM), N2 physisorption, energy dispersive spectrometry (EDS), X-ray photoelectron spectroscopy (XPS) and linear sweep voltammetry (LSV). Then it was applied as an air cathode in a MFC. All the results demonstrated that this non-precious composite with high catalytic activity for oxygen reduction was an excellent alternative to the prohibitive Pt/C catalyst for practical applications in MFCs.
Experimental
Catalyst synthesis
The carbon–nitrogen–metal catalyst, P–Fe, was synthesized by pyrolyzing iron incorporated polyaniline under an NH3 atmosphere. Specifically, 3 ml of distilled aniline and 0.1 g of Fe2SO4 were dispersed in 200 ml of 0.5 M H2SO4 solution. The suspension was kept below 5 °C and ammonium persulfate as an oxidant was slowly dripped into the suspension to initiate the polymerization of aniline. After 6 h of polymerization, the sample containing polyaniline and iron was filtered out, pre-leached by 0.5 M H2SO4 solution for 6 h, washed with deionized (DI) water thoroughly to remove unstable species, and dried under vacuum. Finally, the catalyst was pyrolyzed under an NH3 atmosphere at 900 °C for 30 min.Electrode preparation
A graphite felt and a glassy carbon rotating ring disk electrode (RRDE, disk diameter 5.61 mm, Pt ring) were used in this study. The graphite felt was used as the anodes and cathodes in the MFCs while the RRDE was used to study the electrocatalytic activity of the catalysts towards oxygen reduction in LSV. The graphite felt was pretreated by immersing successively in 1 M NaOH for 12 h and then 1 M H2SO4 for 12 h, followed by rinsing with DI water. The RRDE electrode was pretreated successively in 1.0, 0.3 and 0.05 mm alumina slurries for 3 min at each grade, thoroughly rinsed with DI water and sonicated in Milli-Q water for 5 min.13The cathodes used in the MFCs were prepared by coating P–Fe or Pt/C catalysts onto the graphite felt surface using 5% Nafion solutions as the binder.14 In brief, catalysts were firstly mixed and dispersed well in the Nafion and ethanol solution by ultrasonication. Then, the dispersion was coated onto the graphite felt followed by drying at room temperature. The RRDE electrodes were also coated with P–Fe or Pt/C catalysts using the same method to characterize the electrocatalytic activity of the catalysts towards oxygen reduction. All of the P–Fe coated electrodes had a loading of 1 mg cm−2 and the Pt/C coated electrodes had a Pt loading of 0.5 mg cm−2.
MFC setup and operation
Single chambered air cathode MFCs with a total volume of about 100 ml were constructed as previously described.15 Graphite felt with a diameter of 4 cm was used as the anode and graphite felt with a diameter of 7 cm coated by P–Fe or Pt/C was used as the cathode. Titanium wire was used to connect the anode and the cathode. The MFCs were inoculated using a mixed bacterial culture from another MFC in the laboratory, which was originally started up with primary clarifier overflow from the Gaobeidian Wastewater Treatment Plant in Beijing and had been running for over two years.16 MFCs were fed a medium containing 10 mM sodium acetate, 50 mM phosphate buffer solution (Na2HPO4, 4.09 g l−1 and NaH2PO4·H2O, 2.93 g l−1), NH4Cl (0.31 g l−1), KCl (0.13 g l−1), metal salt (12.5 ml l−1) and vitamin (5 ml l−1) solutions.17 The feeding solution had been sparged with high purity nitrogen gas for 30 min to remove the oxygen that is harmful for the electrochemically active bacteria in MFCs. The MFCs were operated in a temperature-controlled room at 30 °C in a fed batch mode and the feeding solutions were refreshed when the voltage dropped below 40 mV. The MFCs were considered to be stable when the maximum voltage output was reproducible. All of the experiments were carried out in duplicate and the average values with standard deviation were obtained.Analyses and calculations
The morphology of the catalyst was examined using a field emission scanning electron microscope (FESEM) (JSM-6701F, JEOL). The chemical elements on the catalyst surface were analyzed using X-ray photoelectron spectroscopy (XPS) (ESCALab, 220i-XL) and energy dispersive spectrometry (EDS) (INCA X-MAX, Oxford Instruments). The specific surface area was measured using the N2-physisorption method (ASAP 2010, Micromeritics).The RRDE was used to investigate the electrocatalytic activity of the P–Fe catalyst towards the oxygen reduction reaction. The linear sweep voltammetry (LSV) was performed using a conventional three-electrode system at a scan rate of 5 mV s−1 and a rotating speed of 1000 rpm in oxygen saturated 0.1 M HClO4. A Pt foil electrode was used as the counter electrode and a saturated calomel electrode (SCE) was used as the reference electrode. The RRDE electrodes coated with catalyst (or bare GC electrode) were used as the working electrodes. Another indicator of catalytic performance for oxygen reduction was the four-electron selectivity of the catalysts, which was evaluated by the yield of hydrogen peroxide, calculated by the following equation:18 H2O2% = 200 × IR/(IR + NID), where IR is the ring current, ID is the disk current and N is the ring collection efficiency (37% as provided by the manufacturer). In the experiments, the ring potential was set to 1.2 V (vs. SCE).
Cell voltages (U) across an external resistance were measured and recorded using a data acquisition system (AD8201H, Ribohua Co., Ltd). Polarization and power density curves were obtained by varying the external resistance (R) from 10 to 2000 Ω. Current (I) was calculated by I = U/R, and power density (P) was normalized to the MFC reactor volume, calculated as P = U × I/reactor volume.
Results and discussion
Physical and chemical characterization of the catalyst
The morphology of this as-prepared catalyst is shown in Fig. 1. These representative FESEM images indicate that this catalyst exhibits a highly microporous structure. Because of the highly microporous structure, the specific surface area of this catalyst measured by the multiple point BET method was as high as 769.8 m2 g−1 and the pore volume reached 0.36 cm3 g−1. For comparison, the catalyst without pyrolysis in NH3 only had a specific surface area of 12.8 m2 g−1 and a pore volume of 0.01 cm3 g−1. The high specific surface area provided a sufficient interface for the oxygen reduction reaction. In addition, previous studies indicate that micropores in carbon–nitrogen–metal catalysts are crucial for catalytic activity towards oxygen reduction because the active sites are hosted in the micropores.19,20 The EDS spectrum shown in Fig. 2 shows the presence of C, N, O and Fe on the surface of this catalyst. Transition metal (Fe or Co) incorporation has been identified to enhance the oxygen reduction reaction activity in carbon–nitrogen–metal catalysts.8 Therefore, this highly microporous catalyst incorporated with Fe was expected to be very active towards oxygen reduction. | ||
| Fig. 1 FESEM images of the P–Fe catalyst. | ||
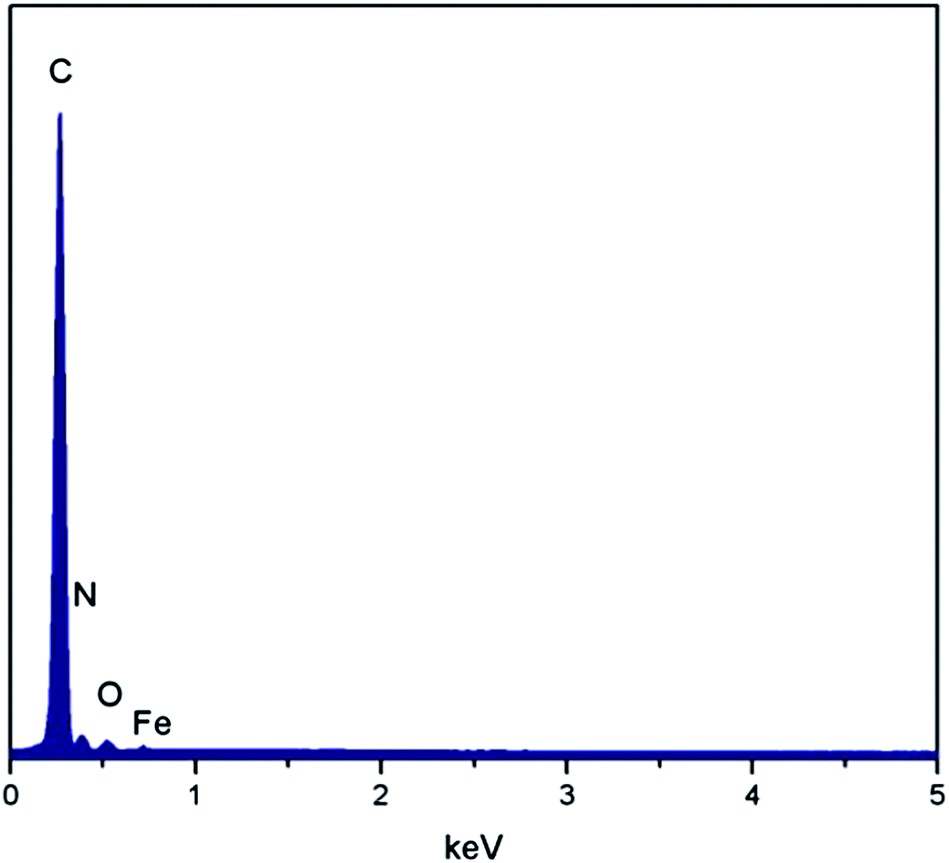 | ||
| Fig. 2 EDS spectrum of the P–Fe catalyst. | ||
The surface chemical composition of the catalyst was characterized using XPS. The general XPS scan of the catalyst (Fig. 3 left) also confirmed the presence of C, N, O and Fe on the surface of this catalyst. Quantitative analysis of this spectrum based on relative sensitivity factors showed that the weight percentage of C, N, O and Fe was 89.46, 7.49, 2.87 and 0.18%, respectively. N content decreased from 17.36% before pyrolysis to 7.49% after heating in NH3. The decrease of surface N content could be due to the gasification of nitrogen-containing fragments by NH3 during pyrolysis. It has been demonstrated that surface N content is the most important factor for the catalytic activity in carbon–nitrogen–metal catalysts; a higher N content leads to a higher density of active sites and consequently better catalytic activity.9,21 Nineteen types of carbon–nitrogen–metal catalysts had been prepared on different carbon supports with surface N content ranging from 0.3 to 2.5%; the catalytic activity is proportional to the surface N content in these catalysts.21 In this study, the surface nitrogen content was much higher than those previously reported catalysts, so this catalyst was expected to exhibit a high electrocatalytic activity for oxygen reduction.
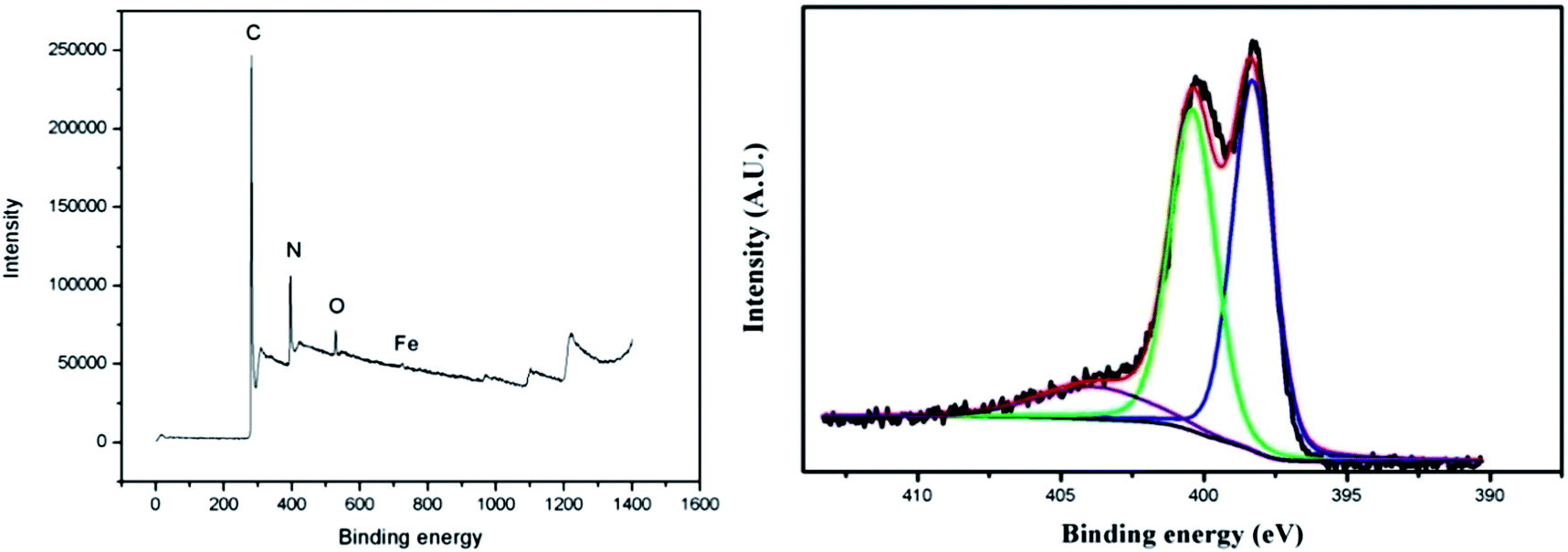 | ||
| Fig. 3 The general XPS scan (left) and the high resolution XPS scan (right) for the N1s core level peak of the P–Fe catalyst. | ||
The electronic states of the surface nitrogen in this catalyst were also analyzed by XPS. The high resolution XPS scan (Fig. 3 right) for the N1s peak of the P–Fe catalyst was deconvoluted into three sets of binding energies originating from pyridinic N (398.3 eV), pyrrolic N (400.7 eV) and oxidized N (403.8 eV).21,22 The relative intensity of the pyridinic N, pyrrolic N and oxidized N was 40.2, 47.7 and 12.1%, respectively. The previous studies have demonstrated that the catalytic sites for the oxygen reduction reaction are made of pyridinic N.9,21,23,24 Therefore, a higher content of pyridinic N was expected to result in more active sites and ultimately, a better electrocatalytic activity.
Electrochemical characterization for oxygen reduction
In order to investigate the electrocatalytic activity of this catalyst towards oxygen reduction, LSV was conducted using a conventional three-electrode system at a scan rate of 5 mV s−1 and a rotating speed of 1000 rpm in oxygen saturated 0.1 M HClO4. For a comparison study, a bare GC electrode and the benchmark Pt/C catalyst were also investigated. As shown in Fig. 4, the bare GC electrode had a quite low onset potential and disk current density, which indicates that GC is a poor electrocatalyst for oxygen reduction. Both the P–Fe and Pt/C electrodes exhibited a high disk current density resulting from the oxygen reduction reaction. Compared with the GC electrode, the P–Fe catalyst showed a much higher onset potential of about 0.92 V (vs. RHE), which was almost the same as that of Pt/C. The performance gap between the P–Fe and the benchmark Pt/C, expressed as half-wave potential difference (ΔE1/2) in this test, was only about 70 mV.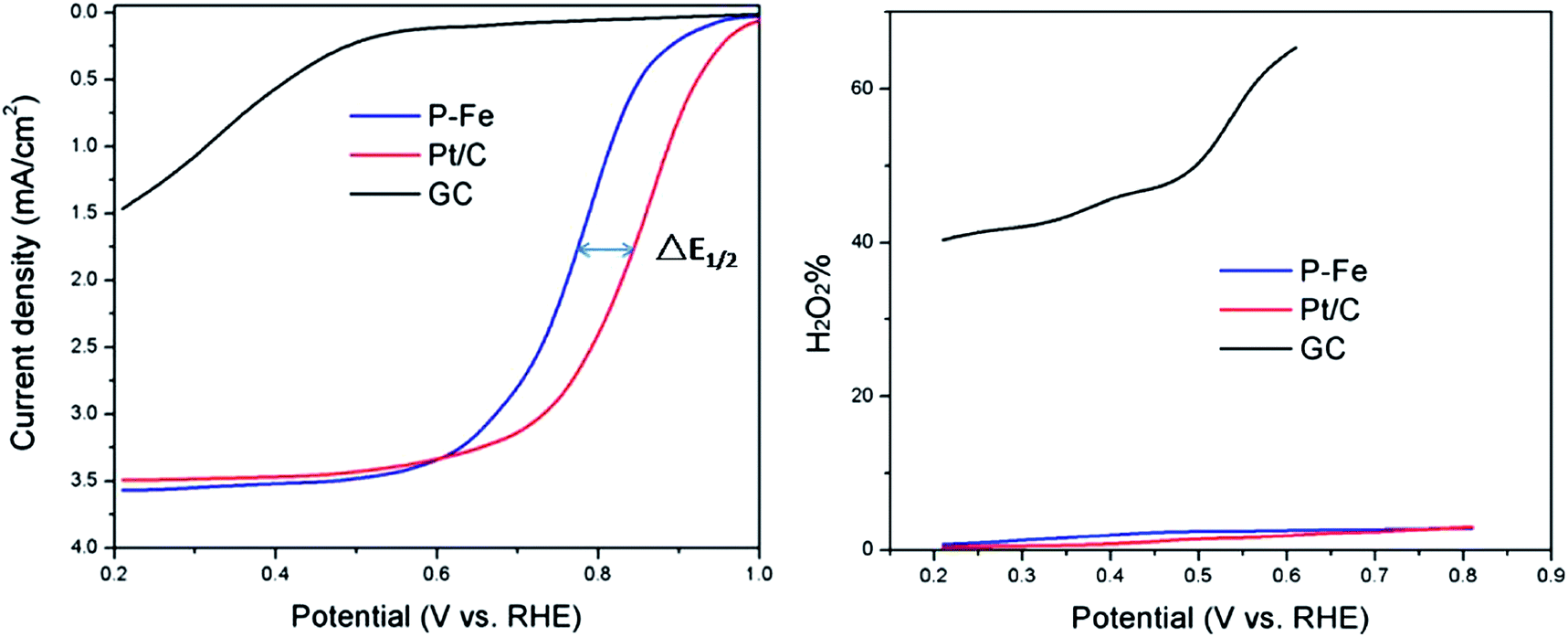 | ||
| Fig. 4 Disk current (left) and H2O2 yield (right) plots during the oxygen reduction reaction in oxygen saturated HClO4 solution (0.1 M) (scan rate: 5 mV s−1; rotating speed: 1000 rpm). | ||
The RRDE study also demonstrated that the P–Fe catalyst is a catalyst with very high selectivity for the four-electron reduction of oxygen. The hydrogen peroxide yield with the P–Fe catalyst was less than 3% in the potential range (Fig. 4 right).
The high onset potential and the high selectivity for the four-electron reduction of oxygen demonstrated that P–Fe is a highly active catalyst towards oxygen reduction. This high electrocatalytic activity could be a result of the highly microporous structure, the presence of iron and the high surface nitrogen content, especially the pyridinic nitrogen. One mechanism for explaining the catalytic activity is that the catalytic sites, FeN4/C and FeN2/C, are created in the micropores during pyrolysis.21,25,26 These catalytic sites hosted in the micropores could effectively lower the activation energy and facilitate the oxygen reduction reaction. It has also been found that these active sites could reduce oxygen predominantly through a four-electron reduction pathway.8,26 In addition, it has been demonstrated that electron-accepting nitrogen atoms can impart a substantially higher positive charge on adjacent carbon atoms to counterbalance the strong electronic affinity of the nitrogen atoms. This nitrogen-induced charge delocalization could change the oxygen adsorption mode from end-on adsorption to side-on adsorption, which can effectively weaken the O–O bonding and improve the oxygen reduction rate.27 Therefore, the key requirements for producing an active carbon–nitrogen–iron catalyst for oxygen reduction are the presence of iron, surface nitrogen and micropores.
Cathode performance in the MFCs
The P–Fe catalyst was used as the cathode to investigate its performance for power generation in MFCs. For comparison, bare graphite felt and the benchmark Pt/C catalyst were also used as controls in the MFCs. When the MFCs became stable in power production, polarization and power density curves were obtained by varying the external resistance. The performance of the P–Fe catalyst in the MFCs was in good agreement with its high electrocatalytic activity in the RRDE study. As shown in Table 1 and Fig. 5, the maximum power density of the MFC with the P–Fe catalyst as the cathode was about 12.56 W m−3, which was significantly higher than the 1.34 W m−3 produced by the MFC with the bare graphite felt electrode and also higher than the 9.92 W m−3 generated by the MFC with the Pt/C catalyst. In addition, the open circuit potential of the P–Fe cathode MFC was also higher than that of the MFCs with the bare graphite felt electrode and the Pt/C catalyst. By calculating the slope of the polarization curves, the internal resistance of the MFCs was determined; results showed that the internal resistance of the MFC using the P–Fe catalyst was 83 Ω, while the internal resistances of the MFCs with the bare graphite felt electrode and the Pt/C catalyst were 209 and 100 Ω, respectively.| Cathodes | Open circuit potential (V) | Internal resistance (Ω) | Maximum power density (W m−3) |
|---|---|---|---|
| Graphite felt | 0.43 | 209 | 1.34 |
| Pt/C | 0.70 | 100 | 9.92 |
| P–Fe | 0.72 | 83 | 12.56 |
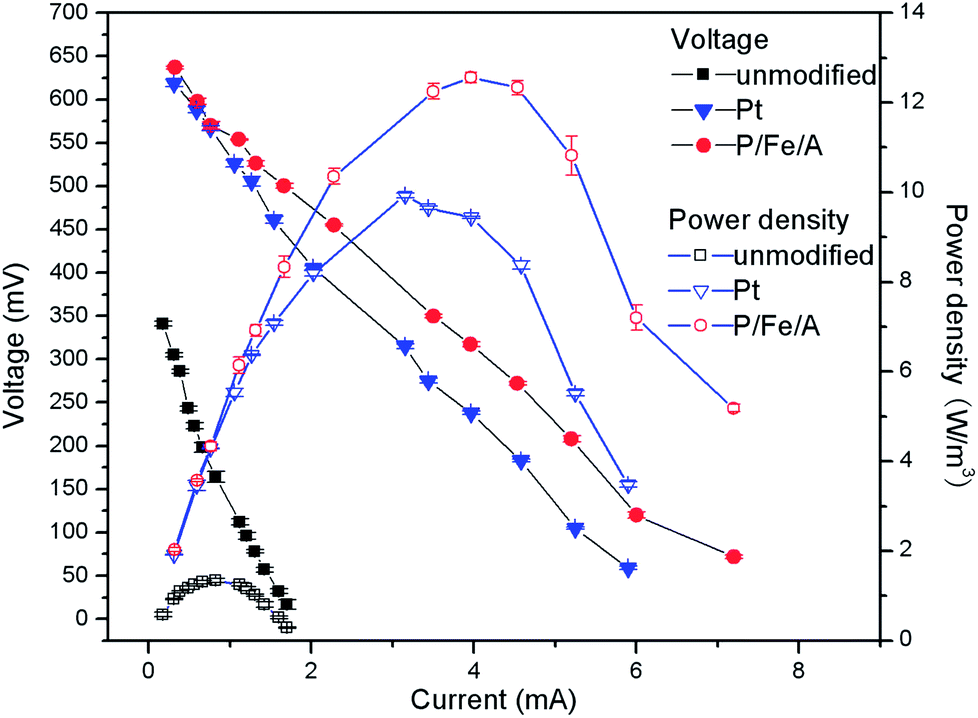 | ||
| Fig. 5 Polarization (solid symbols) and power density (empty symbols) curves of MFCs with different cathodes. | ||
Previous studies have shown that carbon–nitrogen–metal composites exhibit excellent performance as catalysts in the cathodes of H2–O2 fuel cells at 80 °C; the current density of these catalysts can equal that of the Pt/C catalyst at a cell voltage of larger than 0.9 V.8,9 In this study, the performance of the P–Fe catalyst at a mild operating temperature (30 °C) in MFCs was consistent with the good performance of the carbon–nitrogen–metal catalysts in fuel cells, as shown by the high power density and open circuit potential.
The stability of this catalyst was tested by operating the MFCs over a period of 6 months. In the sixth month, the maximum power density of the MFC using this catalyst could still reach 11.98 W m−3, less than a 5% decrease compared to its first month performance. The result demonstrated that the P–Fe catalyst held good stability for oxygen reduction in the MFCs. This was also in line with the report that carbon–nitrogen–metal catalysts displayed excellent stability in a 700 hour fuel cell test; the cell current density in the last 24 hours only dropped by 3% compared with that in the first 24 hours.8
It was important to note that the cost of this catalyst was substantially lower compared with Pt/C, the most popular catalyst used in MFCs. A rough estimation indicated that the cost of the P–Fe produced in the laboratory was less than 1% of the Pt/C cost. Mass production of this catalyst could further bring down the catalyst costs. As mentioned above, the cost of the cathode is the largest part of the cost of an MFC system, which seriously impedes the practical applications of MFCs. Due to the extremely low cost, high catalytic activity towards oxygen reduction and excellent stability, the P–Fe catalyst could be a good alternative to Pt/C in MFCs.
Conclusion
A high-performance electrocatalyst (P–Fe) derived from aniline and iron was synthesized by pyrolyzing iron-incorporated polyaniline under an NH3 atmosphere at 900 °C for 30 min. This catalyst had a highly microporous structure and a high pyridinic nitrogen content on the surface after pyrolyzation. The micropores, pyridinic nitrogen and iron content contributed to the excellent catalytic activity of the P–Fe towards oxygen reduction. The P–Fe catalyst exhibited a high onset potential of about 0.92 V (vs. RHE), which was almost the same as that of Pt/C. The half-wave potential difference between the P–Fe and the benchmark Pt/C was only about 70 mV. The RRDE study demonstrated that the P–Fe is a catalyst with very high selectivity for the four-electron reduction of oxygen. Used as air cathodes in MFCs, the catalyst exhibited a high catalytic activity. The MFC with P–Fe as the catalyst achieved a maximum power density of 12.56 W m−3, significantly higher than the 1.34 W m−3 produced by the MFC with a bare graphite felt electrode and also higher than the 9.92 W m−3 generated by the MFC with the Pt/C catalyst. These results demonstrated that the P–Fe catalyst was an excellent alternative to the cost prohibitive Pt/C catalyst for practical applications.Acknowledgements
This work is supported by a grant from the Environment & Water and Industry Development Council, Singapore (MEWR 651/06/159). We also acknowledge the financial support from the National Natural Science Foundation of China (no. 21176242 & no. 21176026). Tang Xinhua thanks NUS Graduate School for Integrative Sciences and Engineering for a research scholarship support.References
- K. Rabaey, G. Lissens, S. D. Siciliano and W. Verstraete, Biotechnol. Lett., 2003, 25, 1531–1535 CrossRef CAS.
- H. Liu, R. Ramnarayanan and B. E. Logan, Environ. Sci. Technol., 2004, 38, 2281–2285 CrossRef CAS.
- O. Schaetzle, F. Barriere and K. Baronian, Energy Environ. Sci., 2008, 1, 607–620 CAS.
- A. Rinaldi, B. Mecheri, V. Garavaglia, S. Licoccia, P. Di Nardo and E. Traversa, Energy Environ. Sci., 2008, 1, 417–429 CAS.
- D. R. Lovley, Curr. Opin. Biotechnol., 2006, 17, 327–332 CrossRef CAS PubMed.
- A. E. Franks and K. P. Nevin, Energies, 2010, 3, 899–919 CrossRef CAS.
- R. A. Rozendal, H. V. M. Hamelers, K. Rabaey, J. Keller and C. J. N. Buisman, Trends Biotechnol., 2008, 26, 450–459 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS PubMed.
- O. Lefebvre, Z. Tang, M. P. H. Fung, D. H. C. Chua, I. S. Chang and H. Y. Ng, Biosens. Bioelectron., 2012, 31, 164–169 CrossRef CAS PubMed.
- M. Lu, S. Kharkwal, H. Y. Ng and S. F. Y. Li, Biosens. Bioelectron., 2011, 26, 4728–4732 CrossRef CAS PubMed.
- R. Kothandaraman, V. Nallathambi, K. Artyushkova and S. C. Barton, Appl. Catal., B, 2009, 92, 209–216 CrossRef CAS PubMed.
- G. Z. Liu, M. Chockalingham, S. M. Khor, A. L. Gui and J. J. Gooding, Electroanalysis, 2010, 22, 918–926 CrossRef CAS.
- S. Cheng, H. Liu and B. E. Logan, Environ. Sci. Technol., 2006, 40, 364–369 CrossRef CAS.
- P. Wang, B. Lai, H. Li and Z. Du, Bioresour. Technol., 2013, 134, 30–35 CrossRef CAS PubMed.
- X. H. Tang, K. Guo, H. R. Li, Z. W. Du and J. L. Tian, Bioresour. Technol., 2011, 102, 3558–3560 CrossRef CAS PubMed.
- D. R. Lovley and E. J. P. Phillips, Appl. Environ. Microbiol., 1988, 54, 1472–1480 CAS.
- M. B. Vukmirovic, J. Zhang, K. Sasaki, A. U. Nilekar, F. Uribe, M. Mavrikakis and R. R. Adzic, Electrochim. Acta, 2007, 52, 2257–2263 CrossRef CAS PubMed.
- P. H. Matter, L. Zhang and U. S. Ozkan, J. Catal., 2006, 239, 83–96 CrossRef CAS PubMed.
- F. Jaouen, M. Lefevre, J. P. Dodelet and M. Cai, J. Phys. Chem. B, 2006, 110, 5553–5558 CrossRef CAS PubMed.
- F. Jaouen, S. Marcotte, J. P. Dodelet and G. Lindbergh, J. Phys. Chem. B, 2003, 107, 1376–1386 CrossRef CAS.
- H. R. Byon, J. Suntivich and Y. Shao-Horn, Chem. Mater., 2011, 23, 3421–3428 CrossRef CAS.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- E. Proietti, F. Jaouen, M. Lefevre, N. Larouche, J. Tian, J. Herranz and J. P. Dodelet, Nat. Commun., 2011, 2, 416–425 CrossRef PubMed.
- B. Vanwingerden, J. A. R. Vanveen and C. T. J. Mensch, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 65–74 RSC.
- U. I. Koslowski, I. Abs-Wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356–15366 CAS.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |