Palladium(II) induced complete conformational enrichment of the syn isomer of N,N′-bis(4-pyridylformyl)piperazine†
Debakanta Tripathy,
Himansu S. Sahoo,
Venkatachalam Ramkumar and
Dillip Kumar Chand*
Department of Chemistry, IIT Madras, Chennai, India. E-mail: dillip@iitm.ac.in; Fax: +914422574202; Tel: +914422574224
First published on 21st February 2014
Abstract
In the solution state, an equimolar mixture of syn- and anti-conformations is present for the ligand N,N′-bis(4-pyridylformyl)piperazine, L. Combining L with cis-Pd(en)(NO3)2 resulted in the formation of metallomacrocycle cis-[Pd2(en)2(L)2](NO3)4, 1, quantitatively. Coordinated L in complex 1 was found to exist exclusively in the syn-conformation. The equimolar mixture of syn- and anti-conformations was re-established by ethylenediamine induced demetallation of 1.
1. Introduction
External stimuli like light,1 pH,2 guest molecules3 and metal ions4–9 etc. are known to induce conformational changes in flexible ligand systems that have access to two or more conformational states. The role of transition metal ions, as external stimuli, in the process of conformational change has been recognized by exploiting the complexation behavior of potential molecules towards selected metal ions.4–9 Molecules that can undergo conformational changes are useful in many applications such as drug release, information storage, sensor techniques, molecular devices etc.10,11 Recently we reported the conformational behavior of N,N′-bis(3-pyridylformyl)piperazine which exists as an equimolar mixture of the syn- and anti-conformations in the solution state.12 Enrichment of the anti-isomer of the ligand occurs upon complexation with Pd(NO3)2. Herein, we discuss the conformational analysis of a related ligand N,N′-bis(4-pyridylformyl)piperazine (L) that also exists as a mixture of two well defined conformations (i.e. syn and anti, see Fig. 1a) in the solution state. However, conformational enrichment of the syn-isomer of L from an equimolar mixture of the syn- and anti-conformations is established by its complexation with cis-protected Pd(II) (see Fig. 1b). The corresponding decomplexation reaction is also discussed, which shows that the mixture of conformations can be regained by using ethylenediamine as a scavenger. There are reports on guest3 and metal5,7 induced conformational enrichment in which mixtures of two conformers evolve in favor of a preferred one. However, cis-protected Pd(II) induced conformational enrichment of a similar nature has not been reported. This is claimed on the basis of a review published by us containing an exhaustive collection of related coordination cage molecules.13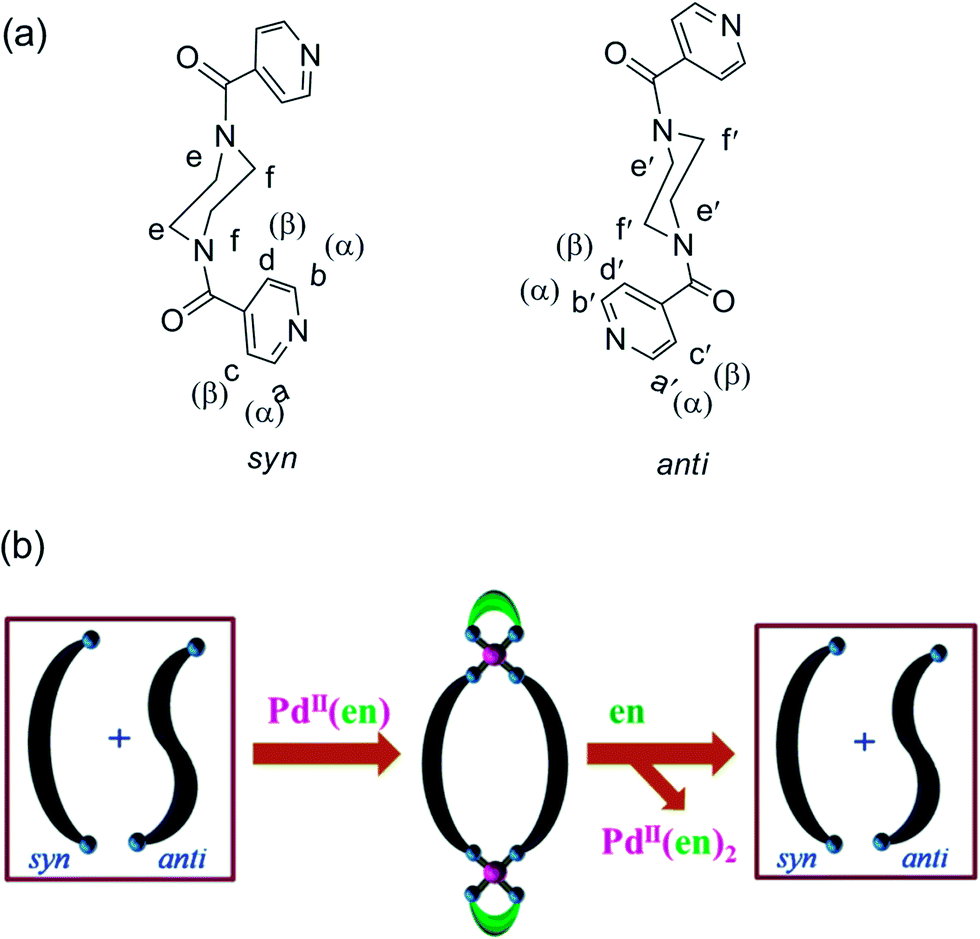 | ||
| Fig. 1 (a) Chemical structure of ligand L (showing the syn- and anti-conformations) and (b) a cartoon diagram showing cis-protected Pd(II) induced conformational enrichment of the syn-isomer of ligand L in step 1 and the re-establishment of the statistical mixture of syn- and anti-isomers in step 2 by decomplexation using ethylenediamine as the scavenger. | ||
2. Results and discussion
Ligand L has previously been employed as a building block for the synthesis of coordination polymers using different metal salts.14–17However, no discrete coordination cages have been prepared using this ligand. We prepared the ligand by slightly modifying reported procedures and these are included in the experimental section.18,19 For the synthesis of the ligand, isonicotinoylchloride hydrochloride was condensed with piperazine in dry DCM in the presence of triethylamine. Usual aqueous workup was performed after completion of the reaction to isolate L as a white powder. Regrettably, NMR spectral data of L is not available in the literature. The 3-pyridyl analogue of ligand L i.e. N,N′-bis(3-pyridylformyl)piperazine, (L′) exists as an equimolar mixture of the syn- and anti-conformations in the solution state.12 Thus ligand L is also expected to behave in a similar way. The behavior of the ligand in solution, as studied from corresponding NMR spectral data, suggested the occurrence of distinct syn- and anti-conformations. This conformational existence is attributed to the restricted rotation imposed by the amide linkages. Like L′, the proton NMR spectrum of L in CDCl3 shows two broad peaks corresponding to the piperazine ring protons and two singlets corresponding to the pyridine α and β protons. The peaks representing the piperazine ring protons are observed at 3.76 and 3.49 ppm, each having a shoulder located at 3.90 and 3.36 ppm, respectively. The singlet at 8.73 corresponds to Ha/Hb/Ha′/Hb′, whereas the singlet at 7.29 is due to Hc/Hd/Hc′/Hd′. The proton NMR spectrum of L in D2O exhibits better resolution (see Fig. 2) indicating a greater degree of restriction imposed by this solvent on the conformational movements in L. In D2O the aliphatic region of the spectrum shows four signals, two of which are singlets (at 3.91 ppm for He and 3.44 ppm for Hf) and the other two are triplets (3.77 ppm for He′ and 3.59 ppm for Hf′). The singlets and triplets correspond to the syn- and anti-conformations, respectively.12 The axial and equatorial protons on a given piperazine carbon are not differentiated, due to the fast conformational flipping between the two possible chair forms. Unlike the two singlets observed in the aromatic region in CDCl3, four doublets are observed in D2O. It is proposed that restricted rotation, due to the partial double bond nature of the bond between the carbonyl carbon and the pyridine γ carbon, is responsible for the resolution observed for these aromatic protons. One set of two very closely spaced doublets, the downfield set, is assigned to the pyα protons i.e. Ha/Ha′ and Hb/Hb′ and the other set to the pyβ protons i.e. Hc/Hc′ and Hd/Hd′. This proposal is further supported by a variable temperature proton NMR study which is discussed in the next paragraph.
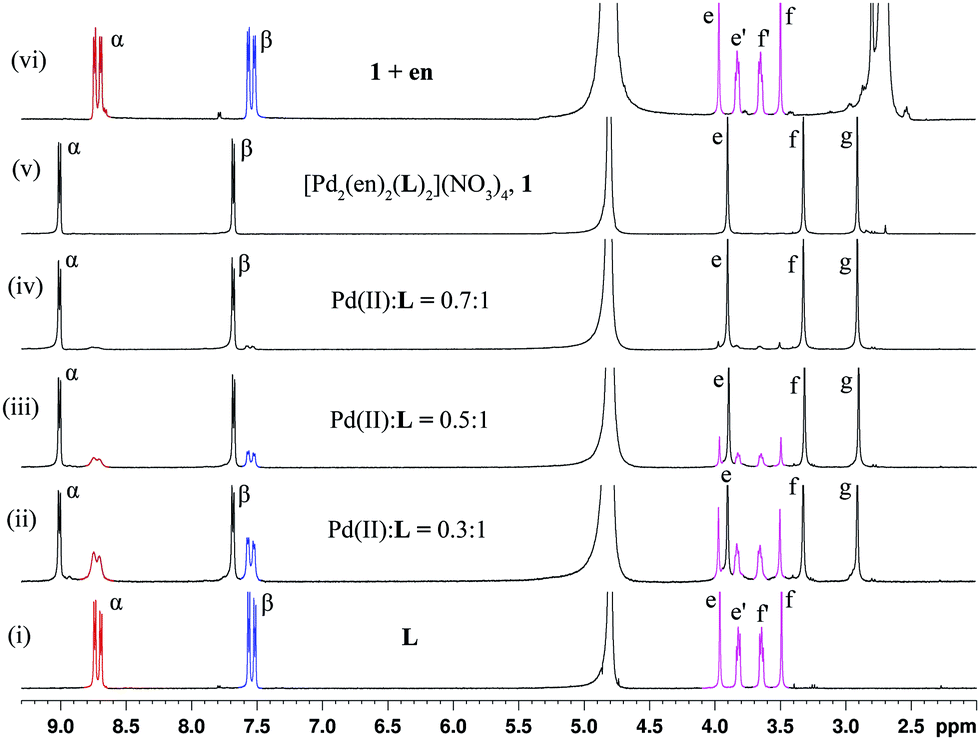 | ||
| Fig. 2 400 MHz 1H NMR spectra in D2O for (i) ligand L (2.7 mg, 0.009 mmol dissolved in 400 μL D2O), (ii)–(v) after the addition of aliquots of cis-Pd(en)(NO3)2 from a stock solution prepared in D2O (stock solution: 2.64 mg, 0.009 mmol in 160 μL). To the sample of L in D2O, the addition of the stock solution of the Pd(II) component was made so that the total amount added was (ii) 50 μL (0.003 mmol), (iii) 80 μL (0.005 mmol), (iv) 120 μL (0.007 mmol), (v) 160 μL (0.009 mmol), and (vi) after the addition of ethylenediamine to 1. The signals shown in colour belong to the free ligand L. For the designation of protons see Fig. 1. | ||
Variable temperature NMR experiments were carried out to investigate the possibility of inter-conversion of the syn- and anti-forms at elevated temperatures. Proton NMR spectroscopy for L was carried out at different temperatures in D2O (ESI†). It was observed that with an increase in temperature the closely spaced doublets corresponding to the pyα protons started broadening. Similar behavior was also observed for the signals representing the pyβ protons. At 60 °C two broad singlets are observed at 9.00 and 7.80 ppm for the pyα and pyβ protons, respectively. Thus, free rotation could happen around the Cpy–CC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds at such an elevated temperature. On the other hand the four signals corresponding to the piperazine ethylene protons remained distinct at 60 °C, however, they appeared like broad singlets. The spectrum after a further increase in temperature to 70 °C also clearly showed four broad singlets in the range 4.40–3.80 ppm. At 80 °C the signals had almost coalesced. This indicates the syn–anti inter-conversion in an NMR time scale due to free rotation around the CCO–Npip bonds. In the case of L′, broadening of the piperazine signals happened at a comparatively lower temperature i.e. at around 40 °C.12
O bonds at such an elevated temperature. On the other hand the four signals corresponding to the piperazine ethylene protons remained distinct at 60 °C, however, they appeared like broad singlets. The spectrum after a further increase in temperature to 70 °C also clearly showed four broad singlets in the range 4.40–3.80 ppm. At 80 °C the signals had almost coalesced. This indicates the syn–anti inter-conversion in an NMR time scale due to free rotation around the CCO–Npip bonds. In the case of L′, broadening of the piperazine signals happened at a comparatively lower temperature i.e. at around 40 °C.12
Single crystals of L suitable for X-ray analysis20 were grown by re-crystallization from hot aqueous solution of the ligand (ESI†). This study aimed to understand the conformational behaviors of L in the solid state. Exclusive existence of the anti-conformation was observed in the single crystal structure of the ligand in this work and has been previously reported by other groups.18 It is worth noting here that the crystals were grown quantitatively upon re-crystallization, thus the fraction of molecules that existed in the syn-conformation in the solution state were converted to the anti-form during the process of crystallization. The single crystals of L were dissolved in D2O and the 1H NMR spectrum was recorded. The spectrum was found to be comparable with that of the pre-crystallized sample. This means that the ligand exists as an equimolar mixture of the syn- and anti-conformations in the solution state, and exclusively in the anti-form in the solid state. The solvation energy is probably sufficient enough to establish a statistical mixture of both of the conformations of comparable free energies. Actually, DFT calculations show a very small energy difference between the syn- and anti-conformations (ESI†). Powder XRD of both the pre-crystallized and the crystallized samples of ligand L are found to be comparable with the simulated data obtained using the single crystal data of L. This indicates the similarity between the conformations of the bulk material and single crystal.
In order to switch the statistical mixture of the syn- and anti-conformations of ligand L to the syn-conformation, it was treated with cis-Pd(en)(NO3)2 to prepare the metallomacrocycle cis-[Pd2(en)2(L)2](NO3)4, 1. In a typical procedure, to the acetonitrile solution of ligand L, cis-Pd(en)(NO3)2 was added and the reaction mixture was stirred for three hours to obtain the desired complex 1 as an off white precipitate. Complex 1 was isolated in a quantitative yield and was characterized by 1H and 13C NMR spectroscopy. All signals were unambiguously assigned without much difficulty due to the very simple nature of the spectra. The analogous complex cis-[Pd2(en)2(L)2](ClO4)4, 1′, was synthesized by combining L with cis-Pd(en)(ClO4)2. The 1H NMR data for 1 and 1′ were found to be exactly the same. The molecular formula of the system was established by carrying out ESI-MS of complex 1′. The ESI-MS spectrum of complex 1′ shows signals at m/z = 341 corresponding to the fragment [1′–3ClO4]3+. The expansion of the envelope of peaks shows the correct isotopic pattern for the above mentioned fragment (ESI†). The 1H NMR spectrum for complex 1 recorded in D2O shows the downfield shifts of the pyridine α protons as well as the β protons due to the palladium–pyridine interaction. The proton NMR signals due to Ha/Ha′ in complex 1 are not distinguishable from those of the Hb/Hb′ protons. Thus, only two doublets are observed in the aromatic region (one for α and the other for β protons). It is worth noting that the region between 3.0 and 4.0 ppm of the NMR spectrum for the complex contains two sharp singlets for the methylene protons of the piperazine moieties. This indicates the highly symmetric nature of complex 1 in the solution state. However, four multiplets corresponding to the methylene protons of the piperazine moieties were observed in the 1H NMR spectrum of the Pd(II) complex containing ligand L′ i.e. [Pd2(L′)4](NO3)4. These multiplets are assigned to the anti-conformation of bound L′ where the piperazine ring is considered flattened. This was finally confirmed by the crystal structure of the complex. It is proposed that the coordinated ligand, L, in 1 exists solely in the syn-conformation. Furthermore, the pyridine rings are probably oriented perpendicular to the corresponding amide planes as suggested by the presence of only two doublets for the pyridine protons and the up-field shift of the peak representing Hf.
The change of the ligand back-bone, from the anti- to the syn-conformation during complexation was monitored by 1H NMR titration (Fig. 2). A sample was prepared by dissolving single crystals of ligand L in D2O in a NMR tube. The required amount of cis-Pd(en)(NO3)2 was added to the sample in a portion-wise manner and the progress of the complexation was monitored by recording 1H NMR after the addition of each aliquot of the metal component. It is observed that both the anti- and syn-conformers are equally consumed during the formation of complex 1. Comparing the 1H NMR spectra obtained here with that of free L shows that the conversion from the anti- to the syn-conformation is very fast during the complexation reactions. After the addition of one equivalent of the metal component the signals corresponding to the uncoordinated L disappeared completely thus giving rise to the complex 1 quantitatively. The region between 4.0 and 3.5 ppm in the 1H NMR spectrum shows only two sharp singlets indicating the complete complexation induced conversion of the mixture of syn- and anti-conformations to only the syn-conformation (Fig. 2). This conformational enrichment proceeded smoothly due to the geometrical commitment required from the ligand for the formation of the binuclear coordination cage.
The existence of the L moiety in the syn-conformation in the metal complex was established by analysing the single crystal X-ray data of 1′. Single crystals suitable for X-ray analysis21 were obtained by slow evaporation of aqueous acetonitrile solution of complex 1′. The complex was crystallized in an orthorhombic crystal system and Cmc21 space group. The asymmetric unit contains half of the molecule and as many as four molecules of 1′ are present in one unit cell. The molecular structure shows that two palladium units are bridged by two ligand moieties to form a binuclear metallomacrocyclic frame-work (Fig. 3). Each of the palladium units are located at the center of slightly distorted square planes comprised of four nitrogens i.e. two from one ethylenediamine unit and the other two from two pyridine units provided by each ligand strand. Two perchlorate ions are found per Pd(II) in line with the requirement of charge balance. Both palladium units are separated by a distance of 12.8 Å. All of the bond lengths and bond angles are in the expected range for the palladium complexes. More importantly the coordinated ligand strands exist in the syn-form in the complex as speculated for the solution state by NMR studies, where the pyridine rings are proposed to be oriented perpendicular to the amide planes. The piperazine moieties are frozen in the chair form. The solution and solid state conformations of the complex are thus found to be the same.
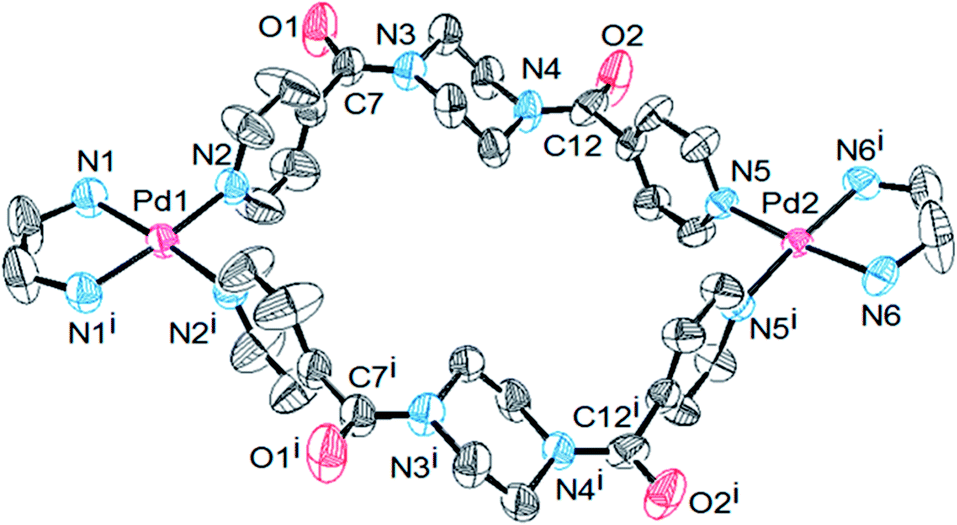 | ||
| Fig. 3 ORTEP diagram of the molecular structure of complexed cation 1′, showing the chair conformation of the piperazine rings and the syn-conformation of the coordinated ligand L (hydrogens, anions and solvent molecules are excluded for clarity; thermal ellipsoids are shown at the 50% probability level). The molecule possesses a plane of symmetry passing through both of the palladium atoms, which bisects the molecule. | ||
The enthalpy of formation of complex 1, comprising the syn-form of ligand L, was estimated by DFT calculations using the B3LYP-LANL2DZ basis sets as included in the Gaussian 09 package. The imaginary isomeric complex 2, comprising the anti-form of ligand L, was also considered using similar calculations. Among the two complexes, complex 1 was found to be more stable by 3.28 kcal mol−1. As can be seen from the energy minimized structure of the isomeric complex 2, the piperazine ring must adopt the energetically unfavorable boat conformation to provide the coordination vectors of the ligand L in a suitable orientation for complex formation (see ESI†). The comparatively low enthalpy of formation for complex 1 probably gives it an edge over the formation of complex 2.
The equimolar mixture of syn- and anti-isomers was regained by a decomplexation reaction where ethylenediamine was added to a solution of 1 in D2O (Fig. 2). The free ligand was released in the process and the NMR spectra of the sample showed the existence of the anti- and syn-conformations of ligand L (Fig. 2).
3. Experimental
3.1. Materials and methods
Isonicotinoyl chloride hydrochloride, PdCl2, AgClO4 were obtained from Aldrich, whereas AgNO3, ethylenediamine and all of the common solvents were obtained from Spectrochem, India, and were used as such without further purification unless specified. Deuterated solvents were obtained from Aldrich and Cambridge Isotope Laboratories. 1H and 13C NMR spectral data were obtained using a Bruker 400 MHz FT NMR spectrometer in D2O and DMSO-d6, using TMS in CDCl3 as an external reference. The ESI mass spectra were obtained using a Micromass Q-TOF mass spectrometer.CAUTION: perchlorate salts are potentially explosive and special care should be taken while using on a big scale. However, no such situation was encountered during the present work.
3.2. Synthesis of ligand, L
Isonicotinoyl chloride hydrochloride (0.814 g, 4.57 mmol) was taken in a 100 mL round bottomed flask and 15 mL of CH2Cl2 was added to it under a nitrogen atmosphere. The suspension was stirred vigorously for 10 minutes followed by the addition of piperazine solution (0.173 g, 2.0 mmol) dissolved in 20 mL of CH2Cl2. Triethylamine (1.2 mL) was added dropwise for about 30 minutes at 0–5 °C. The mixture was stirred at room temperature for 24 hours under a nitrogen atmosphere. To this mixture, NaHCO3 solution (10% w/v) was added slowly to neutralize the acid until the evolution of CO2 had ceased. The organic layer was washed with distilled water, separated and dried over sodium sulfate. Complete evaporation of the solvent gave L as an off white solid (0.504 g, 85%). Colorless crystals were obtained from a hot aqueous solution of the resultant off white solid. Mp 559 K. 1H NMR (CDCl3, 400 MHz, 293 K): δ = 8.73 (s, 4H, pyα), 7.30 (s, 4H, pyβ), 3.91 (bs, 4H, Hpip), 3.74 (bs, 4H, Hpip), 3.50 (bs, 4H, Hpip), 3.36 (bs, 4H, Hpip) ppm. 13C NMR (CDCl3, 100 MHz, 293 K): δ = 168.07, 150.56, 142.54, 121.12, 47.19, 42.14 ppm. ESI-MS (CHCl3): m/z 297 (100%), [(L + H)]+. 1H NMR (D2O, 400 MHz, 293 K): δ = 8.69 and 8.65 (d, J = 4.8 Hz, and d, J = 4.8 Hz, 4H, Ha/a′ and Hb/b′), 7.52 and 7.48 (d, J = 5.2 Hz and d, J = 4.8 Hz, 4H, Hc/c′ and Hd/d′), 3.98 (s, 2H, He), 3.84 (t, 2H, He′), 3.66 (t, 2H, Hf′), 3.51 (s, 2H, Hf). Anal. calcd for C16H16N4O2: C, 64.85; H, 5.44; N, 18.91%. Found: C, 64.67; H, 5.32; N, 18.98%.3.3. Synthesis of complex [Pd2(en)2(L)2](NO3)4, 1
To a solution of ligand L (0.029 g, 0.10 mmol) in 5 mL of acetonitrile, was added cis-[Pd(en)(NO3)2] (0.030 g, 0.10 mmol). The reaction mixture was stirred at room temperature for 3 hours. The resulting precipitate was isolated by centrifugation and was dried under reduced pressure to obtain 1 as a white solid (0.050 g, yield 85%). Mp 451 K. 1H NMR (D2O, 400 MHz, 293 K): δ = 8.94 (d, J = 6.0 Hz, 8H, pyα), 7.60 (d, J = 6.0 Hz, 8H, pyβ), 3.82 (s, 8H, He), 3.24 (s, 8H, Hf), 2.83 (s, 8H, CH2(en)) ppm. 13C NMR (δ, D2O, 100 MHz, 293 K): 167.74, 152.74, 145.99, 124.63, 46.94, 46.82, 41.78 ppm. Anal. calcd for C36H48N16O16Pd2·2H2O: C, 35.74; H, 4.33; N, 18.53%. Found: C, 35.53; H, 3.98; N, 18.21%.3.4. Synthesis of complex [Pd2(en)2(L)2](ClO4)4, 1′
To a solution of Pd(en)Cl2 (32 mg, 0.135 mmol) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile–water mixture (4 mL), was added AgClO4 (0.060 mg, 0.290 mmol). The resulting reaction mixture was heated at 75 °C for 1 h and then centrifuged to remove the precipitated AgCl. To the clear yellow-colored solution was added ligand L (39.9 mg, 0.135 mmol), and the reaction mixture was stirred at room temperature for about 4 h. The colorless solution obtained was evaporated by standing at room temperature, followed by drying under reduced pressure, to obtain 1′ as an off white solid. Yield: 77.6 mg (87%). Mp 483 K (decomposed). 1H NMR (D2O, 400 MHz, 293 K): δ = 8.86 (d, J = 6.4 Hz, 8H, pyα), 7.54 (d, J = 6.8 Hz, 8H, pyβ), 3.75 (s, 8H, He), 3.19 (s, 8H, Hf), 2.75 (s, 8H, CH2(en)) ppm. ESI-MS: m/z = 341 corresponding to [1′–3ClO4]3+. Anal. calcd for C36H52Cl4N12O22Pd2·2H2O: C, 31.80; H, 3.86; N, 12.36%. Found: C, 31.59; H, 3.62; N, 12.05%.
:1 acetonitrile–water mixture (4 mL), was added AgClO4 (0.060 mg, 0.290 mmol). The resulting reaction mixture was heated at 75 °C for 1 h and then centrifuged to remove the precipitated AgCl. To the clear yellow-colored solution was added ligand L (39.9 mg, 0.135 mmol), and the reaction mixture was stirred at room temperature for about 4 h. The colorless solution obtained was evaporated by standing at room temperature, followed by drying under reduced pressure, to obtain 1′ as an off white solid. Yield: 77.6 mg (87%). Mp 483 K (decomposed). 1H NMR (D2O, 400 MHz, 293 K): δ = 8.86 (d, J = 6.4 Hz, 8H, pyα), 7.54 (d, J = 6.8 Hz, 8H, pyβ), 3.75 (s, 8H, He), 3.19 (s, 8H, Hf), 2.75 (s, 8H, CH2(en)) ppm. ESI-MS: m/z = 341 corresponding to [1′–3ClO4]3+. Anal. calcd for C36H52Cl4N12O22Pd2·2H2O: C, 31.80; H, 3.86; N, 12.36%. Found: C, 31.59; H, 3.62; N, 12.05%.
3.5. Crystallographic data collection and refinement
X-ray data was collected using a Bruker AXS Kappa ApexII CCD diffractometer equipped with graphite monochromated Mo(Kα) (λ = 0.7107 Å) radiation. The crystal was fixed at the tip of the glass fiber, was mounted on the Goniometer head and was optically centered. The automatic cell determination routine, with 32 frames at three different orientations of the detector, was employed to collect reflections, and the APEXII-SAINT program (Bruker, 2004) was used for finding the unit cell parameters.22 A 4-fold redundancy per reflection was utilized for achieving good absorption correction using a multiscan procedure. In addition to absorption, Lorentz polarization and decay correction were applied during data reduction. The SADABS program (Bruker 2004) was used for absorption correction using a multiscan procedure. The structures were solved by direct methods using SHELXL-97, (Sheldrick, 2008)23 and refined by full-matrix least-squares techniques using the APEXII (Bruker, 2004) computer program. All hydrogen atoms were fixed at chemically meaningful positions, and a riding model refinement was applied. Molecular graphics were generated using Ortep 3.2 version.4. Conclusions
In conclusion an equimolar mixture of syn- and anti-conformations is present in the solution state of a bidentate ligand, due to restricted C–N bond rotation around the amide linkages. Conformational enrichment of the statistical mixture of conformers is demonstrated by complexation of the ligand with a cis-protected Pd(II) component. Furthermore, the re-establishment of the statistical mixture was achieved by a decomplexation method using ethylenediamine as a scavenger for palladium(II).Acknowledgements
The authors thank the Department of Science and Technology, Government of India for financial support (project no. Sr/S1/IC-28/2009). DT thanks CSIR, India for a fellowship. The authors gratefully acknowledge the single-crystal X-ray Diffractometer facility funded by IIT Madras.Notes and references
- B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS PubMed.
- R. Yamasaki, A. Tanatani, I. Azumaya and S. Saito, Org. Lett., 2003, 5, 1265–1267 CrossRef CAS PubMed.
- G. Giancane, B. Borovkov, Y. Inoue, S. Conoci and L. Valli, Soft Matter, 2013, 9, 2302–2307 RSC.
- S. Ulrich and J.-M. Lehn, J. Am. Chem. Soc., 2009, 131, 5546–5559 CrossRef CAS PubMed.
- Y. S. Chong, M. D. Smith and K. D. Shimizu, J. Am. Chem. Soc., 2001, 123, 7463–7464 CrossRef CAS.
- H. Yuasa, N. Miyagawa, T. Izumi, M. Nakatani, M. Izumi and H. Hashimoto, Org. Lett., 2004, 6, 1489–1492 CrossRef CAS PubMed.
- J. Clayden, L. Vallverdú, J. Clayton and M. Helliwell, Chem. Commun., 2008, 561–563 RSC.
- S. K. M. Nalluri, J. B. Bultema, E. J. Boekemab and B. J. Ravoo, Chem. Sci., 2011, 2, 2383–2391 RSC.
- S. De, S. Pramanik and M. Schmittel, Dalton Trans., 2013, 42, 15391–15398 RSC.
- M. Shahinpoor and H.-J. Schneider, Intelligent Materials, RSCPublishing, Cambridge, 1st edn, 2008 Search PubMed.
- B. L. Feringa, Molecular Switches, Wiley-VCH, Weinheim, Chichester, 2001 Search PubMed.
- H. S. Sahoo and D. K. Chand, Dalton Trans., 2010, 39, 7223–7225 RSC.
- N. B. Debata, D. Tripathy and D. K. Chand, Coord. Chem. Rev., 2012, 256, 1831–1945 CrossRef CAS PubMed.
- C. M. Rogers, C. Y. Wanga, G. A. Farnum and R. L. LaDuca, Inorg. Chim. Acta, 2013, 403, 78–84 CrossRef CAS PubMed.
- C. Y. Wang, Z. M. Wilseck and R. L. LaDuca, Inorg. Chem., 2011, 50, 8997–9003 CrossRef CAS PubMed.
- C. M. Gandolfo and R. L. LaDuca, Cryst. Growth Des., 2011, 11, 1328–1337 CAS.
- H. Xu, Y. Song and H. Hou, Inorg. Chim. Acta, 2004, 357, 3541–3548 CrossRef CAS PubMed.
- Z. M. Wilseck, C. M. Gandolfo and R. L. LaDuca, Inorg. Chim. Acta, 2010, 363, 3865–3873 CrossRef CAS PubMed.
- C.-F. Ding, M. Zhu, X.-M. Li, S.-S. Zhang, H. Xu and P.-K. Ouyang, Acta Crystallogr., 2005, E61, o1981–o1982 Search PubMed.
- Crystal data for L: C16H16N4O2, M = 296.33, monoclinic, a = 9.1945(6) Å, b = 8.2633(5) Å, c = 9.9539(6) Å, V = 721.15(8) Å3, T = 298(2) K, space group P21/n, Z = 4, 4185 reflections measured, 1211 independent reflections (Rint = 0.0213). The final R1 values were 0.0367 (I >2σ(I)). The final wR(F2) values were 0.1259 (I >2σ(I)). The final R1 values were 0.0466 (all data). The final wR(F2) values were 0.1378 (all data). (CCDC 952097).
- Crystal data for 1': C36H48Cl4N12O23Pd2, M = 1371.46, orthorhombic, a = 18.8926(6) Å, b = 17.6085(6) Å, c = 17.0599(6) Å, V = 5675.3(3) Å3, T = 293(2) K, space group Cmc21, Z = 4, 32532 reflections measured, 5142 independent reflections (Rint = 0.0262). The final R1 values were 0.0405 (I >2σ(I)). The final wR(F2) values were 0.0465 (I >2σ(I)). The final R1 values were 0.1244 (all data). The final wR(F2) values were 0.1341 (all data). (CCDC 967775).
- SAINT, version 7.06a, Bruker AXS Inc., Madison, WI, 2004 Search PubMed.
- G. M. Sheldrick, SHELX-97, Programs for Crystal Structure Solution and Refinement, University of Göttingen, Göttingen, Germany, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, ESI-MS, results of DFT calculations and X-ray data of L, 1 and 1′ are given. CCDC 952097 and 967775. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra47904h |
| This journal is © The Royal Society of Chemistry 2014 |