
Nitrogen-based alternative fuel: an environmentally friendly combustion approach
Abstract
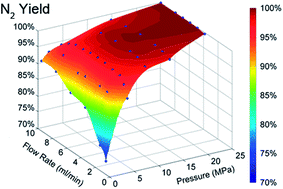
We report here on a continuous combustion of a low carbon nitrogen-based alternative fuel. The investigated fuel, an aqueous solution of urea and ammonium nitrate, consists of common fertilizer commodities. This nonflammable, nontoxic and nonexplosive fuel underwent combustion at a pressure range of 1 to 25 MPa. The pressure was found to greatly affect pollutant levels in the effluent gas. Molecular nitrogen yield was 99.89% at 25 MPa, and the lowest NOx level was 128 mg MJ−1, below the regulation standard for power generation. The alternative fuel described herein is an excellent gas generator, producing an environmentally friendly working fluid consisting of 73.0% H2O, 21.6% N2, and 5.4% CO2. This fuel has the potential to be a sustainable future renewable energy storage medium as well as an energy carrier.
 Please wait while we load your content...
Please wait while we load your content...

