Hydrofluoric acid-induced fluorination and formation of silica nanocapsules for 19F magnetic resonance imaging†
Luís M. F. Lopes and
Laura M. Ilharco*
CQFM – Centro de Química-Física Molecular and IN – Institute of Nanoscience and Nanotechnology, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal. E-mail: lilharco@ist.utl.pt; Fax: +351-218464455; Tel: +351-218419220
First published on 18th February 2014
Abstract
A new, easy and economical method for the production of fluorinated hollow silica nanoparticles using an aqueous hydrofluoric acid solution is reported, and a physico-chemical insight is proposed.
Fluorine-based compounds and materials have found application in a large number of technological domains and give rise to a very active research field, mostly due to their outstanding, and sometimes unexpected, properties.1,2 However, harmful methods of fluorination and expensive fluorinated precursors have hindered their acceptance as practical widespread solutions.
Since, in general, fluorine-based materials are regarded as chemically inert under most biological conditions, a wide range of biomedical applications have been intended, from diagnosis and therapeutic nanocarriers to permanent implants.3–6
Given fluorine's biological rarity and its strong intrinsic magnetic signal, fluorinated compounds and materials labelled with the naturally abundant 19F-isotope have gained relevance in the biomedical imaging field, replacing classical heavy metals7 as contrast agents for Magnetic Resonance Imaging (MRI),8,9 whilst tracer molecules labelled with the radioactive 18F-isotope have been used in Positron Emission Tomography (PET).10,11 Both technologies have emerged as powerful, non-invasive, in vivo imaging tools in oncology, neurology, psychiatry and cardiology.12
In recent years, special attention has been driven towards approaches using fluorinated nanoparticles, mostly silica- and polymer-based, designed for diagnostic imaging tools combined with drug delivery capabilities.13,14 Taking into consideration the biocompatibility, chemical stability and structural control of silica materials versus polymeric matrices, the former tend to be preferred.
Most fluorinated silica nanoparticles found in the literature are obtained either by a sol–gel process involving the condensation of perfluoroalkyl di- or tri-alkoxysilanes,15 or by surface functionalization of preformed silica nanoparticles with perfluoroalkyl alkoxysilanes16,17 or fluoropolymers.18 In some cases, such surface modification has resulted in the improvement of the nanoparticles' solubility, stability and dispersion in different solvents.19 More particular approaches of fluorination have been applied to silica-based materials without nanoparticles, namely direct F2 functionalization,20 and synthesis with hydrofluoric acid (HF)21 or fluoride-containing salts (e.g. KF, NaF, NaBF4 or NH4F), which lead to the formation of [SiF6]2− octahedral species,22 fluorine chemisorption and hydroxyl replacement.23 Other methods include reactive ion etching involving CF4,24 as well as plasma enhanced chemical vapour deposition (PECVD) with SF6, CF4, C2F6, SiF4 or HF.25,26
In the present work, hollow silica nanoparticles with an average external diameter (ϕTEM) of 37 ± 9 nm and with large empty nanocages (Fig. 1A) have been obtained by combining the reverse emulsion method (a mixture of cyclohexane, Triton X-100, hexanol and HF aqueous solution) with the sol–gel reactions of tetraethoxysilane (TEOS), and by subsequent functionalization with 3-aminopropyl-trimethoxysilane (APTS), used in order to increase the particles' stability in biological media. Under these conditions, the silica network formed only at the interface between the aqueous and the cyclohexane phases. From the SEM micrograph (Fig. 1B) it becomes clear that these particles, although not perfectly spherical, exhibit a three-dimensional morphology and a considerable degree of association. This is a drastic change in comparison to the typical morphology of silica nanoparticles prepared by the exact same procedure, but using water instead of HF solution (Fig. 1C): these are larger, spherical, bulk silica nanoparticles with an average diameter (ϕTEM) of 57 ± 5 nm.
 | ||
| Fig. 1 TEM (A) and SEM (B) micrographs of silica nanocapsules fluorinated with a 4%wt HF aqueous solution. For comparison, a TEM micrograph (C) of bulk silica nanoparticles produced by the same procedure, using water instead of HF solution is shown. | ||
The diffuse reflectance infrared Fourier transform (DRIFT) spectrum of these silica nanocapsules (Fig. 2) shows the efficiency of HF as a fluorinating agent: the very strong band at 746 cm−1 is assigned to the νSi–F mode in O4/2SiF species with intermediate geometry between tetrahedral and octahedral, or to the νSi–O–Si mode of relaxed bonds due to the occurrence of strongly polarized Si–F bonds in their vicinity.20 It is so strong that it masks the νsSi–O–Si band expected at ∼800 cm−1. An additional band related to the fluorinated species appears at 991 cm−1, and is assigned to the νsSi–F modes of O2/2SiF2 and O1/2SiF3 tetrahedral species,23 but it is partially overlapped with the stronger νasSi–O–Si band, centred at 1066 cm−1 (Fig. 2B). The weak νSi–OH/νSi–O− band at 937 cm−1 reveals a low content of uncondensed silanol groups or broken siloxane bridges. It attests the efficiency of the silica network condensation, and is confirmed by the low intensity of the νOH broad band centred at ∼3150 cm−1, assigned to hydrogen bonded hydroxyl groups. The small number of free hydroxyl groups present are responsible for the very weak component at a higher wavenumber (3615 cm−1, Fig. 2C).
 | ||
| Fig. 2 (A) DRIFT spectra of fluorinated silica nanocapsules (SiO2–F, gray line) and of non-fluorinated bulk silica nanoparticles (SiO2, black line). The spectra were normalized to the νasSi–O–Si band (1066/1076 cm−1). (B) and (C) Two enhanced regions. | ||
The spectral comparison in Fig. 2 shows that, in the bulk nanoparticles resulting from the same synthesis in the absence of HF, condensation is even more efficient: the bands related to silanol groups are weaker relative to the main silica band. Besides, the maximum of the νasSi–O–Si band is shifted to 1076 cm−1 and its profile is quite distinct. We may correlate these differences to the reaction mechanism and kinetics that give rise to different silica structures. When the aqueous phase within the Triton X-100 micelles is acidified by HF, any TEOS molecules that migrate to the interface are rapidly hydrolyzed and condensed under the catalytic effect of HF, resulting in a silica spherical crown near the interface. When the aqueous phase is neutral, hydrolysis occurs more slowly and only after the addition of ammonia, by a basic mechanism. Under these conditions, the silica nucleation process prevails and the silica network develops, extending to all of the micelle cavities, resulting in bulk nanoparticles.27 The silica structure will be readdressed below, in a more quantitative treatment.
The other features in the spectra of Fig. 2 could be assigned to the surface modifier (APTS), since residual Triton X-100 was removed by a repeated washing procedure. The contribution of APTS bands to the bulk nanoparticles spectrum is not significant, which is not surprising given the much higher relative amount of silica network present. The series of weak bands above 3000 cm−1, overlapped with the broad and very weak νO–H band (Fig. 2C), is assigned to νNH2 modes of APTS, involved in hydrogen bonding with other amine or hydroxyl groups of neighbouring particles. The corresponding scissors mode appears at 1606 and 1512 cm−1 (Fig. 2B). These bands confirm the amine functionalization by the surface modifier, and the different hydrogen bonding interactions between silanol and/or surface amine groups of neighbouring particles. The ρCH3 mode of APTS (which appears as a very strong band at ∼817 cm−1 in the spectrum of the pure compound) is absent, showing that the methoxy groups of APTS have been hydrolyzed. The proposed band assignments are summarized in Table S1 (ESI†).
The interparticle interactions suggested by the DRIFT spectra may justify the association effect observed in the TEM and SEM micrographs (Fig. 1), and have been corroborated by dynamic light scattering (DLS) measurements in deionised water. In fact, for the bulk nanoparticles, a bimodal size distribution was obtained with average diameters of 74 ± 9 and 161 ± 18 nm, corresponding to the hydrodynamic diameters of the individual particles and of the aggregates, respectively. In contrast, for the nanocapsules, only a broad single distribution was obtained, with an average diameter of 507 ± 24 nm, which shows a higher degree of aggregation. The fluorinated nanocapsules and their interactions are schematically represented in Fig. 3.
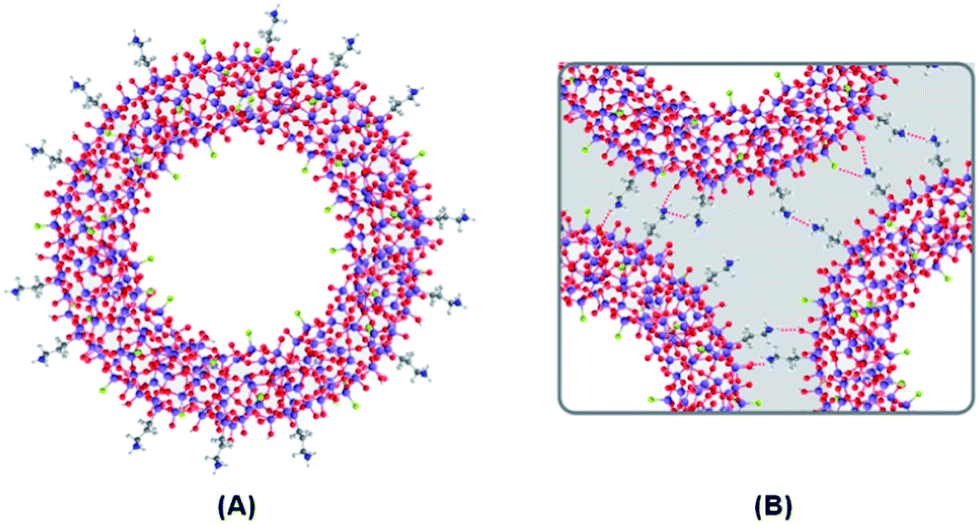 | ||
| Fig. 3 Schematic representation of a fluorinated silica nanocapsule (A) and the interparticle interactions responsible for their association (B). Color code: purple – Si; red – O; gray – C; light gray – H; blue – N; green – F. | ||
The potential of HF as a fluorinating agent was used in a different strategy, consisting of the post-synthesis fluorination of the bulk silica nanoparticles described above. Although the silica dissolution by this acidic source was expected, the results were surprising. By analyzing the differences between the TEM images of these particles before (Fig. 4B) and after (4C) the addition of an 8%wt HF solution, it becomes clear that partial silica destruction of the nanoparticles is promoted by HF, rendering rough surfaces. However, similar to the observations of the fluorinated hollow silica nanoparticles, this process was accompanied by the introduction of fluorine atoms into the silica network, as proved by the appearance of the νSi–F band at 741 cm−1 in the DRIFT spectrum (Fig. 4A) and by the detection of fluorine (at ∼0.7 keV) in the EDS spectrum (Fig. 4D).
 | ||
| Fig. 4 (A) Comparison between the DRIFT spectra of bulk (SiO2, black line) and post-synthesis HF fluorinated (SiO2–post F, gray line) silica nanoparticles, normalized to the νasSi–O–Si band; TEM images of bulk SiO2 (B) and of SiO2–post F (C) nanoparticles; EDS spectrum of SiO2–post F (D). | ||
The breaking of some siloxane bridges induced by the silica dissolution process gives rise to an increase in the number of silanol groups, shown in the DRIFT spectrum by the enhanced relative intensities of the νSi–OH/νSi–O− (at 955 cm−1) and vO–H bands. The broadening of the characteristic νasSi–O–Si band suggests structural modifications of the silica network. The proposed band assignments of the DRIFT spectra of these silica particles are also included in Table S1.†
The silica structure was quantitatively analyzed by deconvolution of the νasSi–O–Si band (860–1300 cm−1) in a sum of Gaussian components. The relevant results are summarized in Table 1.
| Sample | Wavenumber/cm−1a | % (SiO)6b | |||||||
|---|---|---|---|---|---|---|---|---|---|
| νSi–OH/O− | νSi–F | νasSi–O–Si | ωCH2 (APTS) | ||||||
| TO6 | TO4 | LO4 | LO6 | ||||||
| a Relative areas in % – in brackets. TO6 (or TO4) and LO6 (or LO4) correspond to the transverse and longitudinal optic components of the six (or four) membered siloxane rings (SiO)6 (or (SiO)4).b % (SiO)6 = 100 × [A(TO6) + A(TO4)]/A(νasSi–O–Si), where A represents the area of the component between brackets. | |||||||||
| SiO2 | 903 | 951 | — | 1066 | 1076 | 1150 | 1210 | — | 51 |
| (0.5) | (5.9) | — | (42.8) | (8.9) | (37.2) | (4.7) | — | ||
| SiO2–F | 899 | 937 | 991 | 1030 | 1064 | 1127 | 1229 | 1197 | 13 |
| (1.4) | (6.3) | (2.5) | (5.7) | (37.4) | (31.1) | (4.8) | (10.8) | ||
| SiO2–post F | 920 | 959 | 995 | 1067 | 1078 | 1149 | 1212 | — | 55 |
| (2.6) | (5.8) | (0.7) | (43.4) | (0.9) | (40.4) | (6.2) | — | ||
These results were interpreted, taking into account that the elementary SiO4 units of the silica network are mostly arranged in four-membered (SiO)4 and six-membered (SiO)6 siloxane rings, reflected in the spectra by partially overlapped bands corresponding to the νasSi–O–Si mode of each type of ring, and, in each case, split into a pair of longitudinal optic (LO)/transverse optic (TO) components.28 The relative areas of these components (Table 1) confirm that the fluorinated silica nanocapsules have a completely different silica structure from the bulk particles. In the nanocapsules there is a large predominance of four-membered siloxane rings (87%) and a large splitting between the optic components of the six-membered rings (199 cm−1), suggesting a more porous silica structure. Additionally, a component at 1197 cm−1 was clearly retrieved, assigned to the wagging mode of CH2 groups of the surface modifier, and a significant relative intensity was obtained for the Si–F stretching mode. On the other hand, the silica structure of the bulk particles has approximately the same proportion of four- and six-membered siloxane rings, with a slight increase of the latter by post-synthesis fluorination, possibly due to breaking of some siloxane bridges in the more tensioned four-membered rings. As a result, a larger proportion of dangling Si–O bonds appear, responsible for the increase in the relative intensity of the νSi–OH/O− bands (from a total of 6.4 to 8.4%).
In summary, we have demonstrated that HF may act not only as a catalyst for sol–gel processes but also as a fluorinating agent, enabling the synthesis of fluorinated hollow silica nanoparticles by a simple, inexpensive and safer method compared to others. This approach is an unusual and significant step towards the replacement of harmful fluorination methods and expensive precursors. Although most of these particles remain aggregated in deionised water due to the high affinity of functional groups on their surface, a three-dimensional morphology prevails. A successful use of HF as a post-synthesis fluorinating agent of bulk silica nanoparticles was also demonstrated. The resulting nanocapsules could be potentially used for high drug loading transport and delivery, and simultaneously for 19F MRI tracking. In fact, in contrast to the perfluorocarbon compounds (PFCs), these highly fluorinated silica nanoparticles are chemically stable, biologically innocuous and water soluble; besides, the size of the individual particles (below 100 nm) is adequate for intracellular labels, whereas the aggregates may be useful as blood pool agents.29,30
Acknowledgements
Luís Manuel Figueiredo Lopes acknowledges Fundação para a Ciência e a Tecnologia (FCT) for financial support (PhD grant BD/62616/2009).Notes and references
- Fluorine and Health: Molecular Imaging, Biomedical Materials and Pharmaceuticals, ed. A. Tressaud and G. Haufe, Elsevier, Hungary, 2008 Search PubMed.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC.
- J. G. Riess and M. P. Krafft, Biomaterials, 1998, 19, 1529 CrossRef CAS.
- J. G. Riess, Curr. Opin. Colloid Interface Sci., 2009, 14, 294 CrossRef CAS PubMed.
- M. M. Bailey, C. M. Mahoney, K. E. Dempah, J. M. Davis, M. L. Becker, S. Khondee, E. J. Munson and C. Berkland, Macromol. Rapid Commun., 2010, 31, 87 CrossRef CAS PubMed.
- B. D. Ratner, A. S. Hoffman, F. J. Schoen and J. E. Lemons, Biomaterials Science: An Introduction to Materials in Medicine, Academic Press, Canada, 3rd edn, 2013 Search PubMed.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133 CrossRef CAS PubMed.
- M. Srinivas, P. A. Morel, L. A. Ernst, D. H. Laidlaw and E. T. Ahrens, Magn. Reson. Med., 2007, 58, 725 CrossRef CAS PubMed.
- K. Tanaka, N. Kitamura, K. Nakab and Y. Chujo, Chem. Commun., 2008, 6176 RSC.
- R. Schirrmacher, C. Wängler and E. Schirrmacher, Mini-Rev. Org. Chem., 2007, 4, 317 CrossRef CAS.
- G. E. Smith, H. L. Sladen, S. C. G. Biagini and P. J. Blower, Dalton Trans., 2011, 40, 6196 RSC.
- M. J. Goette, A. H. Schmieder, T. A. Williams, J. S. Allen, J. K. Keupp, G. M. Lanza, S. A. Wickline and S. D. Caruthers, J. Cardiovasc. Magn. Reson., 2013, 15(Suppl1), O83 CrossRef.
- J. L. Vivero-Escoto, R. C. Huxford-Phillips and W. Lin, Chem. Soc. Rev., 2012, 41, 2673 RSC.
- E. B. Gyenge, X. Darphin, A. Wirth, U. Pieles, H. Walt, M. Bredell and C. Maake, J. Nanobiotechnol., 2011, 9, 32 CrossRef CAS PubMed.
- H. Sawada, A. Sasaki, K. Sasazawa, K.-I. Toriba, H. Kakehi, M. Miura and N. Isu, Polym. Adv. Technol., 2008, 19, 419 CrossRef CAS PubMed.
- F. Pardal, V. Lapinte and J.-J. Robin, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4617 CrossRef CAS PubMed.
- Y.-C. Sheen, Y.-C. Huang, C.-S. Liao, H.-Y. Chou and F.-C. Chang, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1984 CrossRef CAS PubMed.
- X. Cui, S. Zhong, J. Yan, C. Wang, H. Zhang and H. Wang, Colloids Surf., A, 2010, 360, 41 CrossRef CAS PubMed.
- H. Sawada, T. Narumi, A. Kajiwara, K. Ueno and K. Hamazaki, Colloid Polym. Sci., 2006, 284, 551 CAS.
- E. Lataste, A. Demourgues, H. Leclerc, J.-M. Goupil, A. Vimont, E. Durand, C. Labrugère, H. Benalla and A. Tressaud, J. Phys. Chem. C, 2008, 112, 10943 CAS.
- L. M. F. Lopes, M. N. Kopylovich, A. L. Pombeiro and L. M. Ilharco, J. Phys. Chem. B, 2012, 116, 1189 CrossRef CAS PubMed.
- G. Hartmeyer, C. Marichal, B. Lebeau, P. Caullet and J. Hernandez, J. Phys. Chem. C, 2007, 111, 6634 CAS.
- G. Hartmeyer, C. Marichal, B. Lebeau, S. Rigolet, P. Caullet and J. Hernandez, J. Phys. Chem. C, 2007, 111, 9066 CAS.
- W. Coburn and M. Chen, J. Appl. Phys., 1980, 51, 3134 CrossRef PubMed.
- R. E. Youngman and S. Sen, J. Non-Cryst. Solids, 2004, 349, 10 CrossRef CAS PubMed.
- M. Yoshimaru, S. Koizumi and K. Shimokawa, J. Vac. Sci. Technol., A, 1997, 15, 2915 CAS.
- L. Song, X. Ge, M. Wang and Z. Zhang, J. Non-Cryst. Solids, 2006, 352, 2230 CrossRef CAS PubMed.
- A. Fidalgo and L. M. Ilharco, Chem. - Eur. J., 2004, 10, 392 CrossRef CAS PubMed.
- M. Srinivas, A. Heerschap, E. T. Ahrens, C. G. Figdor and I. J. M. de Vries, Trends Biotechnol., 2010, 28, 363 CrossRef CAS PubMed.
- Y. B. Yu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 646 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and synthesis procedures as well as the characterization techniques are included. Table S1 contains the assignments of the DRIFT spectra of all of the synthesized silica nanoparticles. See DOI: 10.1039/c3ra47842d |
| This journal is © The Royal Society of Chemistry 2014 |