DOI:
10.1039/C3RA47829G
(Paper)
RSC Adv., 2014,
4, 11003-11011
Assembly of evenly distributed Au nanoparticles on thiolated reduced graphene oxide as an active and robust catalyst for hydrogenation of 4-nitroarenes
Received
20th December 2013
, Accepted 5th February 2014
First published on 7th February 2014
Abstract
Thiol-functionalized reduced graphene oxide (SRG) was prepared by coupling the carboxyl groups on the pre-carboxyl-functionalized graphene oxide (GO) with cysteamines through amide bonds. The as-prepared SRG can function both as a support and reducing agent for the formation of Au/SRG nanohybrids with evenly distributed Au nanoparticles on RGO. The thiol linkage on the RGO can act as anchor for Au nanoparticles and due to the existence of the strong interaction between them, the agglomeration of Au nanoparticles can be significantly impeded. The step-by-step assembly process of the Au/SRG nanohybrid was monitored and the products obtained were characterized by Fourier transform infrared spectroscopy (FT-IR), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), X-ray absorption near-edge spectroscopy (XANES), atomic force microscopy (AFM), powder X-ray diffraction (XRD), transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM). The as-prepared Au/SRG nanohybrid showed superior catalytic performance for the hydrogenation of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP). It is expected that the method for the preparation of the Au/SRG nanohybrids can also be applied to the preparations of other RGO-based nanohybrid materials which may find a variety of interesting applications.
1. Introduction
Gold was considered for a long time to be catalytically inactive.1 However, ever since the pioneering work by Bond, Hutchings, Haruta, Prati and Rossi, the catalysis based on Au nanoparticles has attracted extensive research interest.2–5 Gold nanoparticles have been reported to be very active catalysts for a variety of chemical transformations, such as diverse selective oxidations of hydrocarbons,6–8 selective hydrogenation,9–12 low-temperature CO oxidation,13,14 additions to multiple C–C bonds15 and so on. Since Au nanoparticles easily aggregate due to their high surface energy, they are usually anchored on a variety of supports for heterogeneous catalysis, including polymers, metal oxides, carbon materials, etc.16–21 Although there is still much controversy concerning the nature of the active sites of gold nanoparticle catalysts, it has been accepted that the size, dispersion level of the nanoparticles, the interaction between Au nanoparticles and the support are the key parameters determining the performance of the supported Au nanoparticles.
Carbon-based nanomaterials such as carbon nanotubes, graphene and mesoporous carbon are important supports for the metal nanoparticles in heterogeneous catalysis.22–26 Among them, graphene, a novel one-atom-thick two-dimensional graphitic carbon system, has attracted particular research interest due to its high surface area, excellent mechanical, thermal and electrical properties.27–34 Although the anchoring of metal nanoparticles on the pristine graphene is difficult, reduced graphene oxide (RGO) which contains different oxygen groups directly bound to the carbon skeleton of a two-dimensional graphene-derived backbone is an ideal support for anchoring the metal nanoparticles. The surface oxygen functionalities in RGO can serve as reactive sites for the direct nucleation and growth of metal nanoparticles on the RGO surface. However, due to the unevenly distribution of these oxygen functionalities at the surface, the dispersion of the thus prepared metal nanoparticles at the graphene surface are not even.35 To achieve a better distribution of metal nanoparticles, both non-covalent and covalent methods have been applied in the modification of the RGO surface. For example, polymers like PVP, PDDA and so on have been used to modify the graphene surface through electrostatic forces for deposition of Ag, Au and other noble metal nanoparticles.36,37 Chemical modifications on the RGO surface have also been carried out by different reaction types, e.g., electrophilic substitution, nucleophilic substitution, condensation and addition.38–40 The covalent functionalizations on RGO allow the introduction of a wide range of functional groups at the surface which can provide stronger bonding to metal nanoparticles by organic linkers and also prevent the agglomeration of the nanoparticles. For example, it has been reported that thiol group on the support can boost up the interaction between support and Au nanoparticles due to the good stability of the Au–S bond.
In this manuscript, a step-by-step method has been developed to prepare evenly distributed Au nanoparticles at surface thiol-functionalized RGO. Thiol-functionalized RGO was prepared by coupling the carboxyl group on the pre-carboxyl-functionalized graphene oxide surface with the amine group in cysteamine by the condensation agent N,N-dicyclohexylcarbodiimide (DCC). Although this coupling method to form the thiol functionalized surface has been widely used on carbon nanotubes,41,42 it application in the preparation of thiol functionalized RGO surface is limited.43 The as-formed thiol functionality on the surface of RGO can further be utilized as a reducing agent to reduce AuCl4− and the existence of the strong Au–S interaction results in the formation of well-dispersed Au nanoparticles at RGO surface. The as-prepared Au/SRG nanohybrid shows superior catalytic performance for the selective hydrogenation of 4-nitrophenol (4-NP) to the corresponding 4-aminophenol (4-AP). Moreover, the catalyst can easily be separated from the reaction mixture through phase separation and simple filtration for recycling. The step-by-step method in the preparation of Au/SRG nanohybrid is expected to be applied to the preparations of other RGO-supported noble metal nanoparticles with good distribution, uniform particle size and superior catalytic activity.
2. Experimental section
2.1. Materials
Graphite flake (99.8%, 325 mesh) was provides by Alfa. Chloroacetic acid and cysteamine were provided by Aladdin. N,N-Dicyclohexylcarbodiimide (DCC) and anhydrous N,N-dimethylformamide (DMF) were purchased from Sigma Aldrich. All the reagents were used without further purifications.
2.2. Preparations
Graphene oxide (GO). GO was prepared from graphite flake by a modified Hummers method.44 In brief, the graphite flake and NaNO3 were added to cooled 98 wt% H2SO4 under stirring, followed by a gradual addition of KMnO4. The mixture was continuously stirring at room temperature for 5 days. After that, the reaction was diluted by slow addition of H2O and terminated by addition of 30% H2O2 solution. The product was collected by centrifugation and washed repeatedly with diluted HCl. The resultant GO was suspended in water to give a brown dispersion and subjected to dialysis to completely remove the residual salts and acids.
Carboxyl-functionalized graphene oxide (GO-COOH). GO-COOH was prepared following a modified method reported by Sun et al.45 1.2 g of NaOH and 1.0 g of chloroacetic acid were added to a GO suspension. The mixture was then sonicated for 3 h to convert the hydroxyl groups to carboxyl groups. This was followed by neutralizing the solution. After purifying the products by repeated rinsing and filtrations, the resulting GO-COOH was collected for further use.
Thiol-functionalized reduced graphene oxide (SRG). In the thiolation reaction, the carboxyl groups in GO was coupled to the amido groups of cysteamine through amidation. An anhydrous DMF solution containing GO-COOH was mixed with 1 M DCC and a stoichiometric amount of cysteamine in ice-bath. The mixture was sonicated for 1 h and then stirred for 24 h under a nitrogen atmosphere. The resultant SRG was washed with ethanol, filtered and then dried.
Au decorated thiol-functionalized reduced graphene oxide (Au/SRG). 50 μL of 1 wt% HAuCl4 aqueous solution was added to 20 mL of SRG dispersion solution under stirring. After 1 h, the resulting product was collected by centrifugation and washed several times with pure water.
Thiol-capped Au nanoparticles. The thiol-capped Au nanoparticles were prepared according to a literature method.46 100 mL of 0.10 M cysteamine was added to 1 mg mL−1 aqueous metal chloride solution (HAuCl4·3H2O, 200 μL). After stirring for 20 min at R.T., 0.01 mL of 10 mM NaBH4 was added, and the mixture was vigorously stirred in the dark. After 30 min, the product was collected by centrifugation, washed with Milli-Q water three times, and finally re-dispersed in 20 mL water. The Au nanoparticles suspension has a light gray color with the maximum absorption at 540 nm.
2.3. Characterizations
Fourier transform infrared spectroscopy were recorded in transmittance mode with a resolution of 4 cm−1 using a Nicolet Nexus 670 FTIR spectrometer. Powder X-ray diffraction (XRD) data were collected using a Bruker D8 Advance X-ray diffractometer (Cu-Kα1 irradiation, λ = 1.5406 Å). Atomic force microscopy (AFM) image was obtained using an Auto-Probe CP/MT scanning probe microscope (XE-100(PSIA)). X-ray photoelectron spectroscopy (XPS) measurements were performed on a PHI Quantum 2000 XPS system with a monochromatic Al Kα source and a charge neutralizer. All the binding energies were referred to the C 1s peak at 284.8 eV of the surface adventitious carbon. Raman spectroscopy was performed using an invia-Reflex Micro-Raman Spectroscopy system (Renishaw Co.) with 532 nm line of an Ar ion laser at room temperature. The X-ray absorption near-edge spectroscopy (XANES) was collected in Shanghai Synchrotron Radiation Facility (SSRF) with a Si (311) double-crystal monochromator in fluorescence mode. The source of BL14W1 is a 38-pole wiggler device with maximum magnetic field 1.2 T and magnet period 80 mm. The storage ring was operated at 3.5 GeV with injection currents of 100 mA. The transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were measured by a JEOL model JEM 2010 EX instrument at an accelerating voltage of 200 kV. The powder particles were supported on a carbon film coated on a 3 mm diameter fine-mesh copper grid. A suspension of the sample in ethanol was sonicated and a drop was dripped on the support film.
2.4. Catalytic experiments
To study the catalytic activity, 20 mg catalyst was dispersed in 20 mL 1 mM of 4-nitrophenol aqueous solution at room temperature. Then a freshly prepared aqueous solution of 0.4 mL 2 M NaBH4 was added. The reduction performing in the at 1 min intervals was monitored by the UV-vis absorption spectra in the range of 250–550 nm.
3. Results and discussion
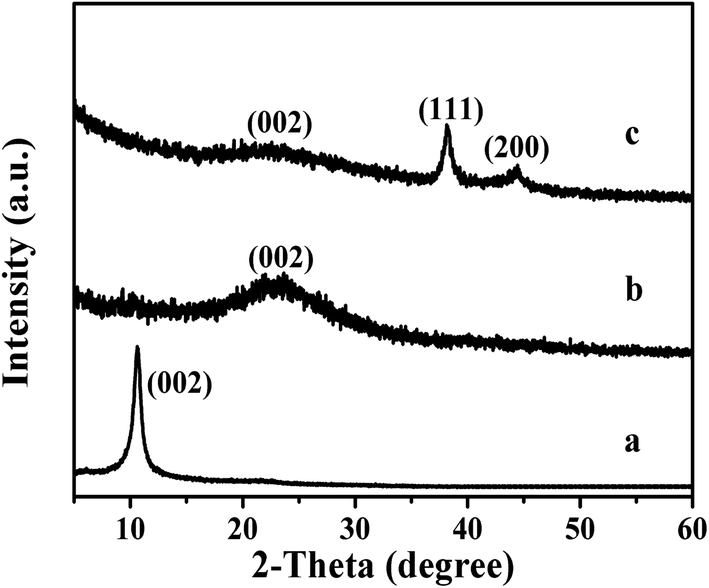
The step-by-step assembly of the Au/SRG nanohybrid through a covalent thiol linkage is shown in Scheme 1. The first step in the preparation involves the chemical oxidation and exfoliation of graphite to graphene oxide (GO) sheets by the modified Hummer's method. The successful formation of exfoliated GO has been confirmed from both the XRD and AFM measurements. As shown in Fig. 1a, after oxidation, the sample shows a well-defined diffraction peak at 2θ = 10.2°, indicative of good layer regularity with a repeating interlayer distance of 0.80 nm. Instead, the original diffraction peak at 2θ of 26.2° corresponding to (002) plane of graphite disappears. The AFM of the sample reveals that the dimension of the resultant GO sample is ca. 2 μm and has a topographic height of about 0.8 nm, indicating that the resultant sample has a single-layer structure (Fig. 2).
 |
| | Scheme 1 Step-by-step assembly of Au/SRG nanohybrid. | |
 |
| | Fig. 1 XRD patterns for (a) GO, (b) SRG and (c) Au/SRG nanohybrid. | |
 |
| | Fig. 2 AFM images and cross-section analysis of GO suspension obtained, deposited on freshly cleaved mica substrates. | |
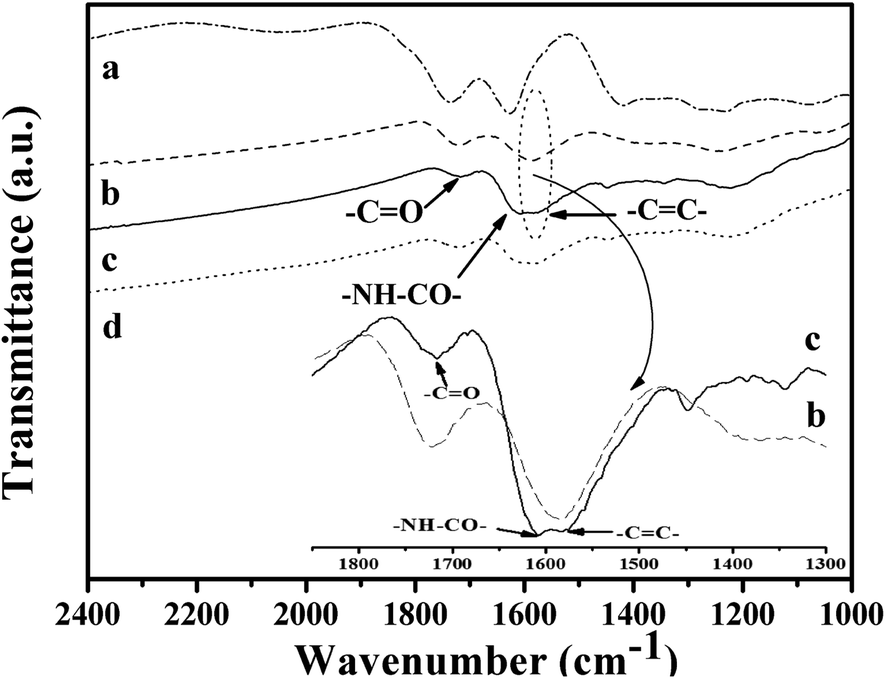
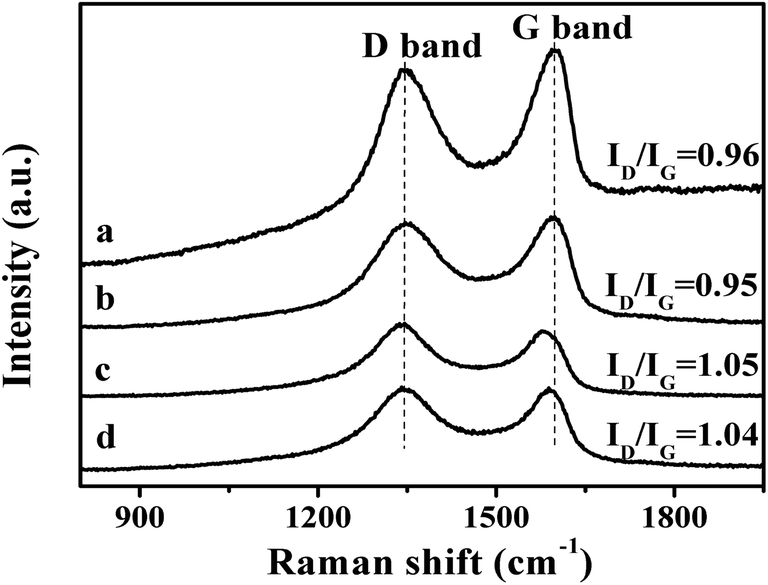
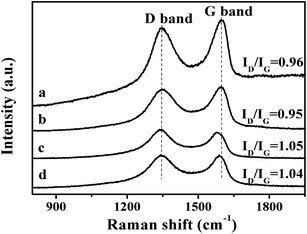
The second step is the carboxylation of the single-layer GO by chloroacetic acid to convert the surface epoxide and hydroxyl groups to carboxylic acid (–COOH) moieties. The FT-IR spectra of the resultant GO-COOH shows peaks at 1712 cm−1 and 1570 cm−1, corresponding to the surface C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from carboxyl group and CC respectively (Fig. 3b).45 As compared to that of the original GO, the peak at 1712 cm−1 significantly increases, while the peaks at 1349 cm−1 and 1055 cm−1 which correspond to the –OH and C–O moieties on the surface decrease obviously (Fig. 3a). This indicates that the surface hydroxyl and epoxide groups on surface of the original GO have been successfully transformed to the carboxyl group during this process. The Raman spectrum of the resultant GO-COOH shows typical D band at 1352 cm−1 and G band at 1592 cm−1, corresponding to the breathing mode of sp2 carbon and graphitic sp2-bonded carbon respectively.47 The intensity ratio of D and G band (ID/IG ratio) for the resultant GO-COOH is determined to be 0.95, comparable to that observed over the original GO (ID/IG = 0.96) (Fig. 4a and b). Since the ratio of ID/IG is an indicator for the extent of defects in the graphene-based materials, the comparable ID/IG ratio observed over GO-COOH and the original GO suggests that the carboxylation process does not induce defects in the resultant GO-COOH.
O from carboxyl group and CC respectively (Fig. 3b).45 As compared to that of the original GO, the peak at 1712 cm−1 significantly increases, while the peaks at 1349 cm−1 and 1055 cm−1 which correspond to the –OH and C–O moieties on the surface decrease obviously (Fig. 3a). This indicates that the surface hydroxyl and epoxide groups on surface of the original GO have been successfully transformed to the carboxyl group during this process. The Raman spectrum of the resultant GO-COOH shows typical D band at 1352 cm−1 and G band at 1592 cm−1, corresponding to the breathing mode of sp2 carbon and graphitic sp2-bonded carbon respectively.47 The intensity ratio of D and G band (ID/IG ratio) for the resultant GO-COOH is determined to be 0.95, comparable to that observed over the original GO (ID/IG = 0.96) (Fig. 4a and b). Since the ratio of ID/IG is an indicator for the extent of defects in the graphene-based materials, the comparable ID/IG ratio observed over GO-COOH and the original GO suggests that the carboxylation process does not induce defects in the resultant GO-COOH.
 |
| | Fig. 3 FTIR spectra of (a) GO, (b) GO-COOH, (c) SRG and (d) Au/SRG nanohybrid. | |
 |
| | Fig. 4 Raman spectra of (a) GO, (b) GO-COOH, (c) SRG and (d) Au/SRG nanohybrid. | |
To prepare the thiol functionalized reduced grapheme oxide, the condensation agent DCC was used to couple the carboxyl group on the GO-COOH surface with the amine group in cysteamine. DCC has been widely used in the formation of polypeptide from the condensation of amino acids. Recently, the application of DCC as the dehydration agent in the formation of amides from amine and carboxyl acids under mild condition has also been reported.41 The FTIR spectrum of the resultant thiol functionalized SRG shows typical amide I band at 1650 cm−1 and amide II band at 1517 cm−1 respectively,43,45 which confirms the formation of the amide bond between the surface carboxyl groups and amine group in cysteamine (Fig. 3c).
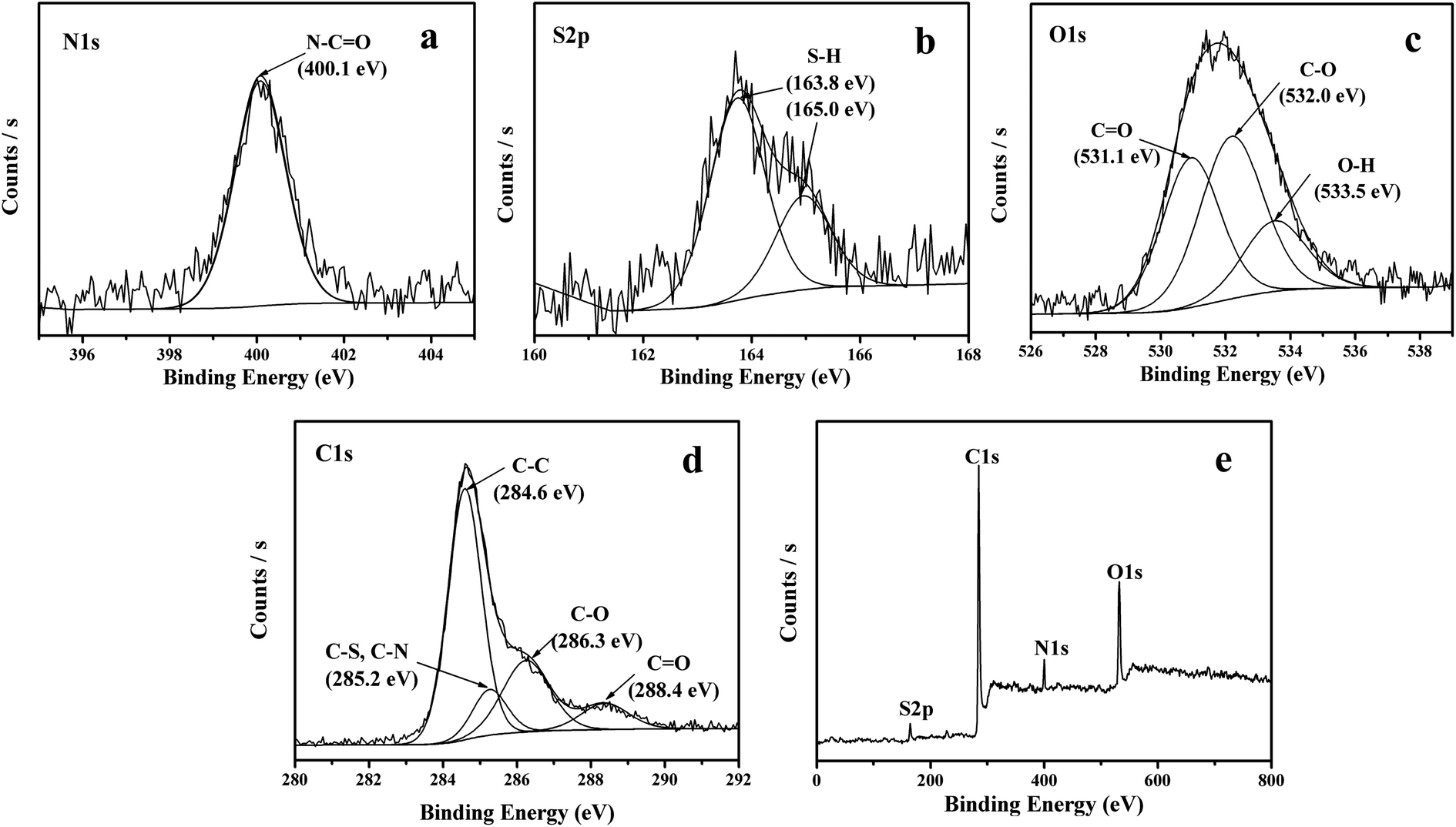
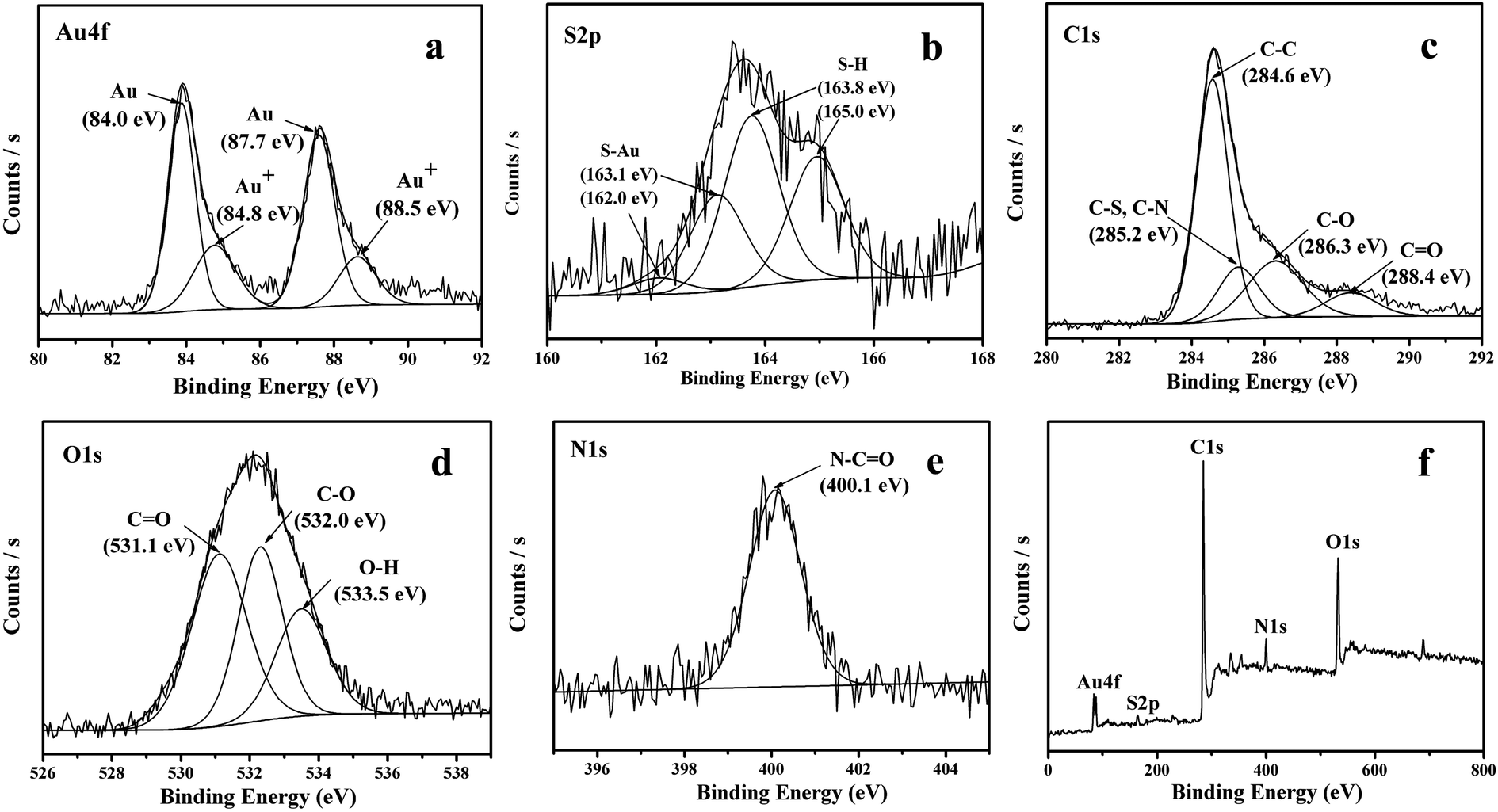
The formation of amide bond has also been confirmed by the XPS spectra. As shown in Fig. 5a, the XPS in the N 1s region shows peaks at 400.1 eV, in agreement with the binding energy of the nitrogen in amide bond.42 The XPS spectra in the S 2p region show peaks at 163.9 and 165 eV, corresponding to the S 2p3/2 and S 2p1/2, a confirmation that the sample is surface functionalized with thiol group (Fig. 5b). The peak in the O 1s region in XPS spectrum can be deconvoluted to 531.1 eV, 532.0 eV and 533.5 eV, which can be assigned to CO, C–O and O–H respectively (Fig. 5c). As compared to original GO, the peak of O–H has dramatically decreased due to carboxylation of the single-layer GO. Similarly, the C 1s peaks can also be deconvoluted to peaks at 284.6, 285.2, 286.3 and 288.4 eV, assigned to CC, C–S or C–N, C–O and CO, respectively (Fig. 5d). The reduction degree of GO defined as the ratio of oxygen-bound carbon content in GO has also been determined based on the XPS results following the equation:
| O-bound C % = (AC–O + AO–CO)/(AC–C + AC–O + AO–CO) × 100% |
where A
C–C, A
C–O, and A
O–CO are the peak areas in the XPS spectra for the sp
2-hybridized (C–C) and O-bound (C–O and O–C
O) carbon, respectively.
48 It is found that after the coupling reaction, the O-bound C content decreases from the original 55% in GO to 27% in SRG, indicating that the coupling reaction also leads to partial reduction of GO. This observation is not surprising since generally the thiol is a reducing agent. The partial reduction of GO by the thiol group would inevitably generate defects in GO. This is also confirmed from the Raman result. The Raman spectra of the thiol functionalized GO show D band at 1352 cm
−1 and G band at 1572 cm
−1 respectively (
Fig. 4c). The
ID/
IG ratio increases from the original 0.95 for GO-COOH to the 1.05 in thiol-functionalized GO, indicating that the coupling process to form the thiol functionalized GO has resulted in the increase of defects.
47 All the above characterizations indicate that cysteamine is grafted on the surface of GO-COOH through the formation of amide linkage.
 |
| | Fig. 5 XPS spectra in (a) N 1s region, (b) S 2p region, (c) O 1s region, (d) C 1s region and (e) survey for SRG. | |
Since that thiols are strong reducing agents and the strong affinity of thiol group to noble metal, especially gold, has been well documented in literature,49,50 the addition of HAuCl4 solution into the as-formed SRG is expected to form the Au nanoparticles on the SRG surface. The XRD pattern of the Au/SRG sample prepared by stirring the SRG suspension in the presence of HAuCl4 for 1 h shows the diffraction peaks at 2θ of 24.3°, 38.1° and 44.3° (Fig. 1c). The peak at 24.3° can be indexed to the (002) crystal face of functionalized graphene oxide, while the other two peaks can be ascribed to (111) and (200) planes of Au in face centered cubic (fcc) structure. This confirms that Au nanoparticles have been successfully obtained on the surface of SRG. The XPS spectra of Au/SRG in Au 4f region show obvious peaks at 87.7 eV and 84.0 eV, attributed to 4f7/2 and 4f5/2 of Au0, and peaks at 88.5 eV and 84.8 eV, corresponding to 4f7/2 and 4f5/2 of Au+ (Fig. 6a). The ratio of Au0 to Au+ is determined to be 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating that most of the Au3+ has been reduced to Au0. As compared to the pristine SRG, the XPS spectrum in the S 2p region can be deconvoluted into two spin-orbit doublets for S–H (163.9 and 165 eV) and S–Au (162 and 163.1 eV), which can be assigned to S bonding to gold nanoparticles and the free thiol groups, respectively (Fig. 6b).42 On the contrary, the XPS spectra of C 1s, O 1s and N 1s do not change obviously, suggesting that the deposition of Au nanoparticles do not significantly influence of the property of the RGO substrate (Fig. 6c–e). Similar results could be obtained from the Au LIII-edge X-ray absorption near-edge spectroscopy (XANES) of Au/SRG (Fig. 7). The characteristic three-peak pattern following the edge jump at about 11.919 keV indicates the presence of the Au0 in a fcc structure.51 As compared with that of Au foil, a slight increase of the first two peaks after the edge jump could be found, which is caused by the minority Au+ that connected with S–H in Au/SRG. The Raman spectrum of the Au/SRG shows D band at 1352 cm−1 and G band at 1592 cm−1 with a ID/IG ratio of 1.04 (Fig. 4d). The comparable ID/IG ratio observed over Au/SRG and pristine SRG (ID/IG = 1.05) indicates that the reduction and deposition of Au nanoparticles does not induce defects on the SRG substrate. The TEM image of Au/SRG shows clearly that Au nanoparticles are evenly deposited on the surfaces of SRG sheets (Fig. 8a). More than 90% of the Au nanoparticles fall in the size range of 15–20 nm with the mean particle diameter determined to be about 17 nm (Fig. 8c), in good agreement with that determined from the XRD result (16 nm). Clear lattice fringes of d = 0.235 nm is observed in the HRTEM image, which matches well with the (111) planes of fcc Au (Fig. 8b). The deposited Au nanoparticles can stand prolonged sonication, indicating that such a step-by-step method can lead to robust and evenly deposited Au nanoparticles on the SRG surface.
:1, indicating that most of the Au3+ has been reduced to Au0. As compared to the pristine SRG, the XPS spectrum in the S 2p region can be deconvoluted into two spin-orbit doublets for S–H (163.9 and 165 eV) and S–Au (162 and 163.1 eV), which can be assigned to S bonding to gold nanoparticles and the free thiol groups, respectively (Fig. 6b).42 On the contrary, the XPS spectra of C 1s, O 1s and N 1s do not change obviously, suggesting that the deposition of Au nanoparticles do not significantly influence of the property of the RGO substrate (Fig. 6c–e). Similar results could be obtained from the Au LIII-edge X-ray absorption near-edge spectroscopy (XANES) of Au/SRG (Fig. 7). The characteristic three-peak pattern following the edge jump at about 11.919 keV indicates the presence of the Au0 in a fcc structure.51 As compared with that of Au foil, a slight increase of the first two peaks after the edge jump could be found, which is caused by the minority Au+ that connected with S–H in Au/SRG. The Raman spectrum of the Au/SRG shows D band at 1352 cm−1 and G band at 1592 cm−1 with a ID/IG ratio of 1.04 (Fig. 4d). The comparable ID/IG ratio observed over Au/SRG and pristine SRG (ID/IG = 1.05) indicates that the reduction and deposition of Au nanoparticles does not induce defects on the SRG substrate. The TEM image of Au/SRG shows clearly that Au nanoparticles are evenly deposited on the surfaces of SRG sheets (Fig. 8a). More than 90% of the Au nanoparticles fall in the size range of 15–20 nm with the mean particle diameter determined to be about 17 nm (Fig. 8c), in good agreement with that determined from the XRD result (16 nm). Clear lattice fringes of d = 0.235 nm is observed in the HRTEM image, which matches well with the (111) planes of fcc Au (Fig. 8b). The deposited Au nanoparticles can stand prolonged sonication, indicating that such a step-by-step method can lead to robust and evenly deposited Au nanoparticles on the SRG surface.
 |
| | Fig. 6 XPS spectra in (a) Au 4f region, (b) S 2p region, (c) C 1s region, (d) O 1s region, (e) N 1s region and (f) survey for Au/SRG nanohybrid. | |
 |
| | Fig. 7 XAFS normalization absorption spectra for Au foil and Au/SRG. | |
 |
| | Fig. 8 Images of Au/SRG nanohybrid (a) low magnification TEM image; (b) HRTEM image; (c) the size distribution of supported Au nanoparticles. | |
Since Au nanoparticles exhibit excellent catalytic activity for the hydrogenation, the catalytic activity of the as-prepared Au/SRG nanohybrid was evaluated by the hydrogenation of 4-nitrophenol (4-NP) in the presence of NaBH4.52 4-Nitrophenol, which is typically found in industrial products and agricultural waste waters, is harmful and hazardous. In addition, the selective hydrogenation of 4-NP to the corresponding 4-aminophenol (4-AP) is known to be one of the fundamental reactions for the synthesis and manufacture of fine and industrial chemicals. Usually this reaction is achieved in the presence of metal nanoparticles, which can catalyze the reaction by facilitating electron relay from the donor BH4− to the acceptor 4-NP to overcome the kinetic barrier.53 Since 4-NP shows a typical absorbance peak at 400 nm, the hydrogenation of 4-NP to 4-AP can decrease the intensity of the peak at 400 nm while a new absorption peak at 300 nm corresponding to 4-AP can develop. As shown in Fig. 9a, when Au/SRG was added into the solution containing 4-NP and NaBH4, the intensity of the strong absorption peak at 400 nm gradually decreased and within 4 min, the whole peak disappeared. In the meantime, a new absorption peak at 300 nm appeared with its intensity increased gradually during this period. This indicates that in the presence of Au/SRG, NaBH4 can hydrogenate 4-NP to 4-AP, and no other by-products have been observed. However, almost no decrease of the peak at 400 nm was observed over pure NaBH4, suggesting that the hydrogenation of 4-NP in the presence of pure NaBH4 can be neglected. Similar hydrogenation of 4-NP in the presence of support-free thiol-capped Au nanoparticles was more slowly and a complete conversion of 4-NP to 4-AP was not achieved even after 5 h reaction (Fig. 9a inset). The low catalytic activity observed over the support-free thiol-capped Au is due to the aggregation of Au nanoparticles in the reaction system. Remarkably, the turnover frequency (TOF) for 4-NP hydrogenation over Au/SRG is calculated to be 60 h−1, much higher than that over polygonal Au nanoparticles (0.4 h−1),54 one of conventional catalyst for 4-NP hydrogenation. Au/SRG also show enhanced activity as compared to Au nanoparticles supported on a variety of supports,55 like PDMAEMA-PS, PNIPAP-b-P4VP, chitosan-coated iron oxide, α-cyclodextrin and graphene oxide, indicating that the thiolated RGO support plays an important role in enhancing the catalytic activity of 4-NP hydrogenation. The increase of the catalytic activity may be due to the following. (i) The thiolated RGO can act as a support for Au nanoparticles and promote the Au-mediated electron transfer from BH4− to 4-NP due to its high electronic conductivity; (ii) a high concentration of 4-NP can be adsorbed on the thiolated RGO via π–π stacking interactions, which is beneficial to the catalytic reaction; (iii) as compared with bare Au nanoparticles, thiolated RGO can offer an environment to prevent the aggregation of Au nanoparticles due to the existence of the Au–S bond, which can help the catalyst to maintain its long term activity.
 |
| | Fig. 9 Time dependent UV-vis absorption spectra for the catalytic hydrogenation of 4-NP by NaBH4 in the presence of (a) Au/SRG nanohybrid and thiol-capped Au nanoparticles (insert). (b) Comparative plots of Ct vs. time for Au/SRG nanohybrid towards the hydrogenation of 4-NP by NaBH4. | |
By assuming the reaction to be pseudo-zero-order56 which is reasonable because the concentrations of 4-nitrophenol and NaBH4 largely exceeded those of the catalysts, the catalytic rate constant (k) could be determined from the curve plotted of the concentration of 4-nitrophenol Ct, i.e., the absorbance versus reaction time (t). As shown in Fig. 9b, a good relationship can be built between Ct and t and the corresponding k calculated from the linear slope was 0.5 mM min−1 for Au/SRG nanohybrid.
The stability of the Au/SRG nanohybrid for the catalytic hydrogenation of 4-NP has also been investigated. As shown in Fig. 10a, Au/SRG nanohybrid shows almost similar catalytic activity for the hydrogenation of 4-NP over five cycles of use. In addition, the catalyst after the reaction shows similar XRD patterns as that of the fresh one, indicating that the metallic state of Au is still kept (Fig. 10b inset). The TEM image of the sample after the catalytic reaction shows that Au with the same size distribution is still evenly dispersed on the surface (Fig. 10b). All these suggest that the Au/SRG nanohybrid is an active, robust and reusable catalyst for hydrogenation of 4-NP.
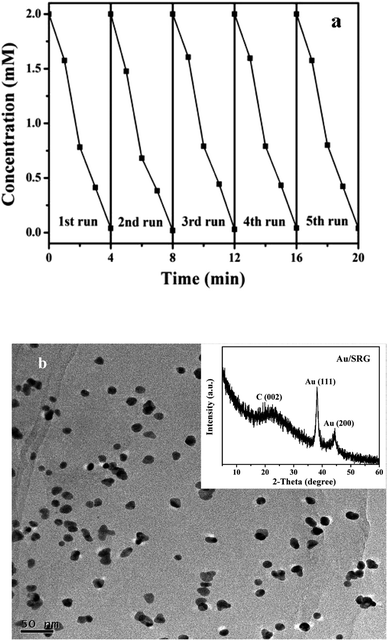 |
| | Fig. 10 (a) Cycling runs in the catalytic hydrogenation of 4-NP over Au/SRG nanohybrid. (b) TEM image and XRD pattern (inset) of Au/SRG nanohybrid after the catalytic cycling runs. | |
4. Conclusions
An efficient method for the preparation of thiol-functionalized RGO has been developed. Evenly dispersed Au nanoparticles on RGO can be obtained by using the thiol-functionalzied RGO as a reducing agent as well as a support. The thiol linkage on the RGO can act as anchor for Au nanoparticles and due to the existence of the strong interaction between them, the agglomeration of Au nanoparticles can be significantly impeded. The as-prepared Au/SRG nanohybrid shows superior catalytic activity and high stability for the selective hydrogenation of 4-NP to the corresponding 4-AP. It is expected that the step-by-step method for the preparation of Au/SRG nanohybrids can also be applied to the preparations of other RGO-based nanohybrid materials which may find a variety of interesting applications.
Acknowledgements
We are grateful to Shanghai Synchrotron Radiation Facility (SSRF) of China for the X-ray absorption near-edge spectroscopy (XANES) measurements at the BL14W1 beamline. The work was supported by 973 Programs (2014CB239303), NSFC (21273035, 21077023) and Specialized Research Fund for the Doctoral Program of Higher Education (20123514110002). Z. Li thanks the Award Program for Minjiang Scholar Professorship for financial support.
References
- H. Schmidbaur, Naturwiss. Rundsch., 1995, 48, 443–451 Search PubMed.
- G. C. Bond, P. A. Sermon, G. Webb, D. A. Buchanan and P. B. Wells, J. Chem. Soc., Chem. Commun., 1973, 444–445 RSC.
- G. J. Hutchings, J. Catal., 1985, 96, 292–295 CrossRef CAS.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552–560 CrossRef CAS.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC.
- A. Abad, C. Almela, A. Corma and H. García, Chem. Commun., 2006, 3178–3180 RSC.
- C. D. Pina, E. Falletta and M. Rossi, Chem. Soc. Rev., 2012, 41, 350–369 RSC.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed.
- M. Pan, H. C. Ham, W. Y. Yu, G. S. Hwang and C. B. Mullins, J. Am. Chem. Soc., 2013, 135, 436–442 CrossRef CAS PubMed.
- M. Pan, Z. D. Pozun, A. J. Brush, G. Henkelman and C. B. Mullins, ChemCatChem, 2012, 4, 1241–1244 CrossRef CAS.
- M. Pan, A. J. Brush, Z. D. Pozun, H. C. Ham, W. Y. Yu, G. Henkelman, G. S. Hwang and C. B. Mullins, Chem. Soc. Rev., 2013, 42, 5002–5013 RSC.
- Y. Maeda, Y. Iizuka and M. Kohyama, J. Am. Chem. Soc., 2013, 135, 906–909 CrossRef CAS PubMed.
- Z. Wu, S. Zhou, H. Zhu, S. Dai and S. H. Overbury, J. Phys. Chem. C, 2009, 113, 3726–3734 CAS.
- M. Bandini, Chem. Soc. Rev., 2011, 40, 1358–1367 RSC.
- H. Yin, H. Tang, D. Wang, Y. Gao and Z. Tang, ACS Nano, 2012, 6, 8288–8297 CrossRef CAS PubMed.
- F. Shi, Q. Zhang, Y. Ma, Y. He and Y. Deng, J. Am. Chem. Soc., 2005, 127, 4182–4183 CrossRef CAS PubMed.
- C. Y. Ma, Z. Mu, J. J. Li, Y. G. Jin, J. Cheng, G. Q. Lu, Z. P. Hao and S. Z. Qiao, J. Am. Chem. Soc., 2010, 132, 2608–2613 CrossRef CAS PubMed.
- Y. Guan and E. J. M. Hensen, Appl. Catal., A, 2009, 361, 49–56 CrossRef CAS PubMed.
- Z. Wang, J. Zhang, Z. Yin, S. Wu, D. Mandler and H. Zhang, Nanoscale, 2012, 4, 2728–2733 RSC.
- Z. Zanolli, R. Leghrib, A. J. Felten, J. J. Pireaux, E. Llobet and J. C. Charlier, ACS Nano, 2011, 5, 4592–4599 CrossRef CAS PubMed.
- L. Chen, K. Yang, H. Liu and X. Wang, Carbon, 2008, 46, 2137–2143 CrossRef CAS PubMed.
- P. V. Kamat, J. Phys. Chem. Lett., 2010, 1, 520–527 CrossRef CAS.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876–1902 CrossRef CAS PubMed.
- X. Xie, J. Long, J. Xu, L. Chen, Y. Wang, Z. Zhang and X. Wang, RSC Adv., 2012, 2, 12438–12446 RSC.
- S. Wang, Q. Zhao, H. Wei, J. Q. Wang, M. Cho, H. S. Cho, O. Terasaki and Y. Wan, J. Am. Chem. Soc., 2013, 135, 11849–11860 CrossRef CAS PubMed.
- S. Pang, Y. Hernandez, X. Feng and K. Müllen, Adv. Mater., 2011, 23, 2779–2795 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich and E. J. Zimney, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin and G. H. B. Dommett, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- W. Wei, T. He, X. Teng, S. Wu, L. Ma, H. Zhang, J. Ma, Y. Yang, H. Chen, Y. Han, H. Sun and L. Huang, Small, 2012, 8, 2271–2276 CrossRef CAS PubMed.
- Y. Kopelevich and P. Esquinazi, Adv. Mater., 2007, 19, 4559–4563 CrossRef CAS.
- T. Schwamb, B. R. Burg, N. C. Schirmer and D. Poulikakos, Nanotechnology, 2009, 20, 405704 CrossRef PubMed.
- C. G. Navarro, R. T. Weitz and A. M. Bittner, Nano Lett., 2007, 7, 3499–3503 CrossRef PubMed.
- Y. B. Zhang, Y. W. Tan and H. L. Stormer, Nature, 2005, 438, 201–204 CrossRef CAS PubMed.
- G. Goncalves, P. A. A. P. Marques, C. M. Granadeiro, H. I. S. Nogueira, M. K. Singh and J. Gracio, Chem. Mater., 2009, 21, 4796–4802 CrossRef CAS.
- Z. Zhang, F. G. Xu, W. S. Yang, M. Y. Guo, X. D. Wang, B. L. Zhang and J. L. Tang, Chem. Commun., 2011, 47, 6440–6442 RSC.
- Y. Fang, S. Guo, C. Zhu, Y. Zhai and E. Wang, Langmuir, 2010, 26, 11277–11282 CrossRef CAS PubMed.
- J. Liu, J. Tang and J. J. Gooding, J. Mater. Chem., 2012, 22, 12435–12452 RSC.
- H. Yao, L. Jin, H. J. Sue, Y. Sumi and R. Nishimura, J. Mater. Chem. A, 2013, 1, 10783–10789 CAS.
- D. Marquardt, F. Beckert, F. Pennetreau, F. Tölle, R. Mülhaupt, O. Riant, S. Hermans, J. Barthel and C. Janiak, Carbon, 2014, 66, 285–294 CrossRef CAS PubMed.
- S. Park, H. R. Kim, J. Kim, C. Jung, C. K. Rhee, K. Kwon and Y. Kim, Carbon, 2011, 49, 487–494 CrossRef CAS PubMed.
- L. Minati, G. Speranza, S. Torrengo, L. Toniutti, C. Migliaresi, D. Maniglio, M. Ferrari and A. Chiasera, Surf. Sci., 2010, 604, 1414–1419 CrossRef CAS PubMed.
- E. S. Orth, J. E. S. Fonsaca, S. H. Domingues, H. Mehl, M. M. Oliveirab and A. J. G. Zarbin, Carbon, 2013, 61, 543–550 CrossRef CAS PubMed.
- A. Lerf, H. Y. He and M. Forster, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- X. M. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. J. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- S. N. Sarangi, A. M. P. Hussain and S. N. Sahu, Appl. Phys. Lett., 2009, 95, 73109 CrossRef PubMed.
- C. K. Chua and M. Pumera, Chem. – Eur. J., 2013, 19, 2005–2011 CrossRef CAS PubMed.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed.
- N. R. Jana and X. Peng, J. Am. Chem. Soc., 2003, 125, 14280–14281 CrossRef CAS PubMed.
- C. Yang, G. An and X. Zhao, J. Mater. Sci.: Mater. Electron., 2013, 24, 3490–3495 CrossRef CAS PubMed.
- P. Zhang, X. Zhou, Y. Tang and T. K. Sham, Langmuir, 2005, 21, 8502–8508 CrossRef CAS PubMed.
- Y. Choi, H. S. Bae, E. Seo, S. Jang, K. H. Park and B. S. Kim, J. Mater. Chem., 2011, 21, 15431–15436 RSC.
- J. Huang, L. M. Zhang, B. Chen, N. Ji, F. H. Chen, Y. Zhang and Z. J. Zhang, Nanoscale, 2010, 2, 2733–2738 RSC.
- M. H. Rashid and T. K. Mandal, Adv. Funct. Mater., 2008, 18, 2261–2271 CrossRef CAS.
- K. Layek, M. L. Kantam, M. Shirai, D. N. Hamane, T. Sasaki and H. Maheswaran, Green Chem., 2012, 14, 3164–3174 RSC.
- Y. M. Zhang, X. Yuan, Y. Wang and Y. Chen, J. Mater. Chem., 2012, 22, 7245–7251 RSC.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.