
A water soluble FRET-based ratiometric chemosensor for Hg(ii) and S2− applicable in living cell staining†
Abstract
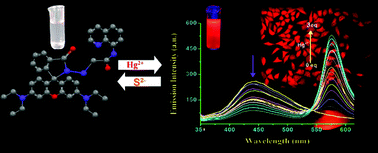
A new highly sensitive and selective Hg(II) probe, 2-(rhodamine-b-hydrazido)-N-(quinolin-8-yl)acetamide (L1) was developed and characterized. L1 specifically binds to Hg(II) in the presence of a large excess of other competing ions with visually observable changes in both electronic and fluorescence spectral behaviour to make possible the naked eye detection of Hg(II) at a very low level (up to 4.5 × 10−7 M) through a fluorescence resonance energy transfer (FRET) process in HEPES buffer (1 mM, pH 7.4; 2% EtOH) at 25 °C. The theoretical and experimental kinetic study also support the binding of Hg(II) ion to induce the opening of the spirolactam ring in L1 for enabling the FRET process. Further studies reveal that the selective dissociation of the L–Hg complex in the presence of sulphide anions to restore the native structure of L1 is also useful in the detection of sulfide anions with a detection limit of a submicromolar range in the same medium of HEPES buffer (1 mM, pH 7.4; 2% EtOH) at 25 °C. L1 could be employed as a FRET based time dependent reversible chemosensor for imaging Hg(II) in living cells and whole bodies, and also could be used as an imaging probe for the detection of sulfide anions in HeLa cells.
- This article is part of the themed collection: Luminescence and photophysical properties of metal complexes
 Please wait while we load your content...
Please wait while we load your content...

