
Diastereoselection during 1,2-addition of 3-bromomethyl-5H-furan-2-one to α-chiral aldehydes mediated by indium in aqueous and organic solvent systems: direct route to obtain optically active α-methylene-γ-butyrolactones†
Abstract
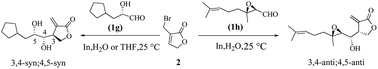
The stereochemical course of indium-promoted allylations to α-chiral aldehydes with 3-bromomethyl-5H-furan-2-one was investigated in anhydrous THF and pure H2O. High levels of 3,4-syn;4,5-syn diastereomers were produced with free hydroxyl derivatives whether in THF or pure H2O, reflecting the promising synthetic potential of this chemistry. This stereo-differentiation was attributed to the strong geometric bias exercised by our allylindium reagent and adherence to a chelation control transition-state alignment. Meanwhile, heightened levels of 3,4-anti;4,5-anti diastereomers were obtained with the rigid aldehydes 1h and 1i.
 Please wait while we load your content...
Please wait while we load your content...

