Metal catalyst free one-pot synthesis of 2-arylbenzimidazoles from α-aroylketene dithioacetals†
Pandi Dhanalakshmi,
Solaimalai Thimmarayaperumal and
Sivakumar Shanmugam*
Department of Organic Chemistry, School of Chemistry, Madurai Kamaraj University, Madurai 625021, India. E-mail: shivazzen@mkuniversity.org
First published on 7th January 2014
Abstract
An efficient green synthetic approach has been developed towards the synthesis of 2-aryl substituted benzimidazoles from α-aroylketene dithioacetals (AKDTAs) 1 and o-phenylenediamine (OPD) 2. The reaction has been achieved in water with a catalytic amount of acetic acid. 2-Arylbenzimidazoles have been synthesized in remarkable yields under both thermal and microwave conditions. The metal catalyst free condition makes this transformation very green, practical and attractive.
Introduction
A great number of biologically active molecules contain the benzimidazole scaffold and especially 2-substituted benzimidazole has been found to be biologically more potent. Their application is further extended as poly(ADP-ribose) phosphorylase inhibitors,1 histamine H4 receptor binders,2 antiparasitic,3 cardiovascular,4 anticancer,5 antimicrobial,6 and antihypertensive6 agents. In addition, benzimidazoles have been found to have antiulcer activity and inodilator applications (Fig. 1).7 The AKDTAs 1 are three carbon synthons, and they are highly functionalized α,β-unsaturated ketones containing both electron withdrawing carbonyl group and electron donating alkylthio substituents well known for their donor–acceptor double bond. The alkylthio group is a very good leaving group and it can be easily replaced by nucleophiles. In several reactions, the AKDTAs 1 behave as α,β-unsaturated carbonyl compounds wherein, depending upon the nucleophile/reaction conditions either 1,2- or 1,4-addition takes place. In acidic media nucleophiles prefer 1,2- addition and it has been extensively used for the synthesis of wide variety of heterocyclic compounds.8 | ||
| Fig. 1 Structures of representative benzimidazole core motif. | ||
Several methods have been reported for the synthesis of 2-substituted benzimidazoles.9–11 Generally, the conventional method involves the reaction of aryl aldehyde/carboxylic acid or their derivatives with 1,2-diamines to afford the benzimidazole at elevated temperature in the presence of strong acids like polyphosphoric acid,12 and mineral acid,13–15 several other catalysts like indium triflate,16 iodine,17 cetylpyridinium bromide18 PEG-40019 (bromodimethyl)sulfonium bromide,20 ammonium acetate,21 cobalt(II) chloride hexahydrate,22 ceric ammonium nitrate23 and enzymatic catalyst lipozyme24 have been used instead of mineral acids for this cyclocondensation. Recently, substituted benzimidazole has been reported by reacting 2 with aryl aldehydes using 4-OMe-TEMPO as the catalyst under aerobic conditions.25 In addition, C–N bond formation via a cross coupling reaction, and direct C–H activation using a transition metal catalyst have also been reported to construct the benzimidazole skeleton.26
The distinctive methods of assembling these valuable heterocycles are highly dependent on using 2 as the precursor. The literature for the other methods to synthesize 2-arylbenzimidazoles have been comprehensively reviewed (Scheme 1).27–32
 | ||
| Scheme 1 Strategies for the synthesis of 2-arylbenzimidazoles. | ||
Water mediated organic synthesis has become one of the most attractive protocols in view of environmental aspects. We now report a conceptually novel, simple and effective metal catalyst free direct cyclocondensation of readily available AKDTAs 1 with 2 in the presence of acetic acid as a catalyst in water to afford 4 in excellent yields. The development of a lab route for the synthesis of benzimidazole 4 under metal catalyst free and eco-friendly conditions is worth considering for its practical approach on larger scale operations. Rao et al. reported trisubstituted pyrrole33 and 3-aroyl coumarin34 by reacting AKDTA with TosMIC and salicylaldehydes, respectively. In continuation of exploring the synthetic potential of AKDTAs 1, we were interested to construct seven membered benzodiazepine derivatives which are pharmacologically and biologically valuable.35–37
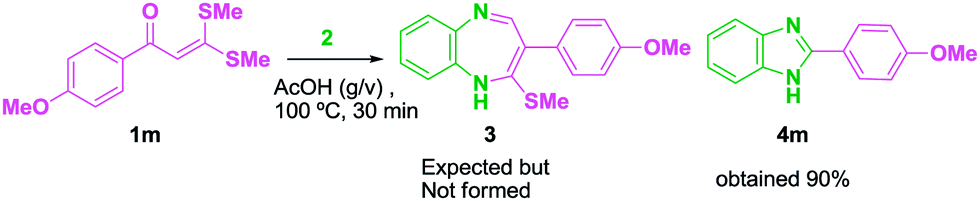
Following the literature,34 a variety of AKDTAs 1 have been synthesized (Table 1). Further, a mixture of AKDTA 1m and 2 was heated in the presence of glacial acetic acid (g/v) at 100 °C for 30 min (Scheme 2). The reaction went smoothly and the crude product was purified by recrystallization with ethanol.
|
|||
|---|---|---|---|
| Entry | Ar | Yielda (%) | |
| a Isolated yield after recrystallization. | |||
| 1 | C6H5 | 1a | 88 |
| 2 | 2-Naphthyl | 1b | 92 |
| 3 | 1-Naphthyl | 1c | 85 |
| 4 | 4-ClC6H4 | 1d | 92 |
| 5 | 4-CH3C6H4 | 1e | 82 |
| 6 | Ferrocenyl | 1f | 85 |
| 7 | Pyrenyl | 1g | 80 |
| 8 | 3-NO2C6H4 | 1h | 80 |
| 9 | 3-OCH3C6H4 | 1i | 85 |
| 10 | 2-FC6H4 | 1j | 84 |
| 11 | 3-CF3C6H4 | 1k | 85 |
| 12 | 4-BrC6H4 | 1l | 80 |
| 13 | 4-OCH3C6H4 | 1m | 80 |
| 14 | 2,4-Cl2C6H3 | 1n | 82 |
| 15 | 3,4-F2C6H3 | 1o | 83 |
| 16 | 3,4-Cl2C6H3 | 1p | 79 |
| 17 | 3-BrC6H4 | 1q | 84 |
| 18 | 2-CF3C6H4 | 1r | 84 |
| 19 | 2-F,5-CF3C6H3 | 1s | 70 |
| 20 | 4-IC6H4 | 1t | 77 |
| 21 | 2-F,4-CF3C6H3 | 1u | 72 |
| 22 | 3-CF3,4-ClC6H3 | 1v | 77 |

 | ||
| Scheme 2 Cyclocondensation between 1m and 2 in AcOH. | ||
The isolated product was well characterized by 1H and 13C NMR spectra. Anticipating the compound to be 3, the 1H NMR spectrum displayed a singlet at δ 12.86 for the aromatic NH-proton and one singlet at δ 3.82 for OMe group, a pair of doublets at δ 7.09 and 8.09 ppm with mutual coupling constant J = 8.8 Hz for the two CH protons of the phenyl group of 1m. Two multiplets were appeared at δ 7.14–7.15 and 7.46–7.59 for the CH aromatic protons of 2. But no peak was noticed for the SMe group and the olefinic proton at δ 2.48 ppm and δ 6.86 ppm. Thus the NMR data do not match with compound 3 but perfectly match with 4m and the mass spectrum [m/z 225 (M + 1)] also confims the formation of benzimidazole. Extensive literature studies revealed that this is the first report to the synthesis of 2-arylbenzimidazole 4 from AKDTA 1. Some of the earlier reports for the synthesis of 2-arylbenzimidazole involve the use of expensive metal catalysts, more reaction time and steps and expensive reagents. The advantages of the present method are that it is metal catalyst free with less reaction time and excellent yields. All of the starting materials of AKDTAs 1 are solids and stable, with high melting points and can be easily prepared with simple reaction techniques. Hence, we report the synthesis of 2-arylbenzimidazole by a conceptually novel method of cyclocondensation between AKDTAs 1 and 2. The cyclocondensation was optimized with 4, which was observed through several reactions between AKDTA 1 and 2 (Table 2).
| Entry | Solvent | Catalyst (mol%) | Temp. (°C) | Time | Yield (%)d |
|---|---|---|---|---|---|
| a Reaction failed to occur.b Unreacted 1 & 2 recovered.c Complex reaction mixture.d Isolated yield.e Reaction performed at microwave (MW, 120 W, 100 °C). | |||||
| 1 | None | None | 100 | 3 h | Nra |
| 2 | None | AcOH (100) | Rt | 72 h | 60b |
| 3 | None | AcOH(100) | 100 | 15 min | 75 |
| 4 | None | AcOH (40) | 100 | 30 min | 87 |
| 5 | None | AcOH (30) | 100 | 30 min | 65b |
| 6 | None | Formic acid (50) | 80 | 3 h | —c |
| 7 | EtOH | None | 90 | 3 h | Nra |
| 8 | EtOH | AcOH (40) | 90 | 3 h | 60b |
| 9 | MeCN | AcOH (40) | 80 | 2 h | 75b |
| 10 | MeCN | Yb(OTf)3 (50) | 80 | 5 h | 65b |
| 11 | DMF | Yb(OTf)3 (50) | 100 | 5 h | 75b |
| 12 | EtOH | p-TsOH (40) | 100 | 30 min | 83 |
| 13 | EtOH | p-TsOH (40, MW)e | 100 | 2 min | 85 |
| 14 | H2O | p-TsOH (MW) | 100 | 2 min | 50 |
| 15 | H2O | H2SO4 (1 M) | 100 | 30 min | —c |
| 16 | H2O | HCl (1 M) | 100 | 30 min | 50b |
| 17 | H2O | AcOH (100) | 100 | 1 h | 87 |
| 18 | H2O | AcOH (40) | Rt | 120 h | 20b |
| 19 | H2O | AcOH (40) | 100 | 2 h | 95 |
Once 4m had been synthesized, several cyclocondensations were tried by variation of reaction conditions (Table 2). The reaction time increased gradually when we reduced the amount of AcOH. The absence of acetic acid did not give the product 4m (Table 2, entry 1). We examined the mild Lewis acid ytterbium(III) trifluoromethanesulfonate (Yb(OTf)3) with two different solvents to afford 4m in 65 and 75% yields (Table 2, entry 10 & 11). With a catalytic amount of p-TsOH in ethanol, the reaction proceeded smoothly in both thermal (30 min) and microwave (MW, 2 min) conditions with good yield (Table 2, entry 12–13). The drawback of this condition is in water/p-TsOH media under microwave condition only 50% of product was observed (Table 2, entry 14). In HCl, the yield was very low and H2SO4 and formic acid media did not give the desired product (Table 2, entry 15–16 & 6). The reaction was performed in ethanol, acetonitrile and water as solvents in the presence of AcOH (40 mol%) at 100 °C. Among the above solvents, a mixture of water–AcOH (40 mol%) gave 4m in maximum yield (95%, Table 2, entry 19). The optimal reaction conditions were a result of 2-(4-methoxyphenyl)-1H-benzo[d]imidazole 4m with 2 in the presence of water–AcOH (40 mol%) at 100 °C for 2 h. Overall, the synthesis of 2-arylbenzimidazoles from AKDTA 1 and 2 proceeds only in the presence of acid medium.
In the next step, variation of the aryl groups was studied and several 2-arylbenzimidazole derivatives 4a–v were synthesized using the optimal reaction conditions (Table 3). Halogen substituted aryls and polyaryls such as 1 and 2-naphthyl, pyrene and ferrocene substituted benzimidazoles were consecutively investigated and the yields were excellent. Some of the final products 4a,c,f–g,j,l and 4t were obtained as pure crystalline products after simple work-up with saturated sodium bicarbonate solution. Later, we carried out the reaction between 1 and 2 under microwave irradiation at 100 °C for 5 min to furnish 4a–v in good to excellent yields (78–90%, Table 3). There is not much difference in the yield of the products, when compared with thermal conditions. Thus the shorter reaction time and good to excellent yields encouraged us to repeat all the reactions under microwave irradiation (Table 3).
|
||||
|---|---|---|---|---|
| Entry | Ar | Yield | ||
| Thermal (%) | MWc (%) | |||
| a All reactions carried out with 1 (1 mmol), 2 (1 mmol), AcOH (40 mol%), water at 100 °C (i) thermal 2 h, (ii) MW 5 min.b Yields after recrystallization.c Isolated yield. | ||||
| 1 | C6H5 | 4a | 95b | 90 |
| 2 | 2-Naphthyl | 4b | 92 | 88 |
| 3 | 1-Naphthyl | 4c | 93b | 87 |
| 4 | 4-ClC6H4 | 4d | 88 | 82 |
| 5 | 4-CH3C6H4 | 4e | 87 | 82 |
| 6 | Ferrocenyl | 4f | 88b | 83 |
| 7 | Pyrenyl | 4g | 90b | 87 |
| 8 | 3-NO2C6H4 | 4h | 89 | 82 |
| 9 | 3-OCH3C6H4 | 4i | 88 | 83 |
| 10 | 2-FC6H4 | 4j | 82b | 79 |
| 11 | 3-CF3C6H4 | 4k | 84 | 80 |
| 12 | 4-BrC6H4 | 4l | 90b | 85 |
| 13 | 4-OCH3C6H4 | 4m | 90 | 88 |
| 14 | 2,4-Cl2C6H3 | 4n | 85 | 81 |
| 15 | 3,4-F2C6H3 | 4o | 84 | 78 |
| 16 | 3,4-Cl2C6H3 | 4p | 83 | 80 |
| 17 | 3-BrC6H4 | 4q | 89 | 84 |
| 18 | 2-CF3C6H4 | 4r | 83 | 79 |
| 19 | 2-F,5-CF3C6H3 | 4s | 80 | 78 |
| 20 | 4-IC6H4 | 4t | 90b | 86 |
| 21 | 2-F,4-CF3C6H3 | 4u | 87 | 82 |
| 22 | 3-CF3,4-ClC6H3 | 4v | 87 | 83 |

There are number of mechanisms envisioned for the formation of 2-arylbenzimidazole 4 from o-phenylenediamine 2 with aldehyde or acid or acid chloride etc. Based on the results, we propose a plausible mechanism for the one-pot synthesis of 4 by the cyclocondensation of AKDTA 1 and 2 (Scheme 3). When the mixture of AKDTA 1 and 2 with catalytic glacial acetic acid was heated at 100 °C, initially 1 is protonated followed by instantaneous nucleophilic addition of an amine group of 2 at C-1 position to give imine(II). This imine formation makes the C-1 position more electron deficient and attracts further nucleophilic addition by another amine group of 2 to afford a five membered heterocycle 2-(2,2-bis(methylthio)vinyl)-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole(III). In order to achieve the aromaticity, elimination of ethynyl(methyl)sulfane 5 by cleavage of the C–C bond of the ketene group and MeSH 6 has occurred to afford 4 in excellent yields. Elimination of 5 and 6 is the driving force for the formation of 4.
 | ||
| Scheme 3 Plausible mechanism for the two component cyclocondensation. | ||
Conclusions
In summary, we have demonstrated the synthesis of substituted arylbenzimidazoles from AKDTAs 1a–v and readily available 2 under mild and green medium in excellent yields for both thermal and MW conditions. It is noteworthy that this methodology is very simple, less time consuming, metal catalyst free, involving eco-friendly solvent and milder reaction conditions. The economical and environmental advantages of this protocol adds practical value for industrial applications.Experimental section
General methods
The melting points reported in this work are uncorrected. Unless stated otherwise, solvents and chemicals were obtained from commercial sources and used without further purification. The 1H and 13C NMR spectra of the new compounds were measured at 300 or 400 MHz in DMSO-d6 and CDCl3. Chemical shifts are reported as δ values (ppm) relative to tetramethylsilane (δ 0.0) as internal standard. Mass spectra were obtained using an electrospray ionization (ESI) mass spectrometer and recorded in positive and negative mode. Infrared spectra were recorded on an FT-IR spectrometer with the major peaks listed. HRMS (ESI-TOF) analyses were recorded on a mass spectrometer. Petroleum ether employed in column chromatographic purification refers to the fraction which boils at 40–60 °C. Microwave reactions have been carried out in a Biotage Microwave Synthesizer.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc = 7:3), the mixture was transferred into a 100 ml beaker containing 50 g of crushed ice and the contents of the beaker were stirred well. A light yellow coloured solid that formed was filtered and washed with water (10 ml × 3). The crude solid was re-crystallized from EtOH to furnish 3.10 g of 1,1-di(methylsulfanyl)-3-(aryl)-1-propen-3-one in 80–92% yield as light yellow colored crystals.
:EtOAc = 7:3), the mixture was transferred into a 100 ml beaker containing 50 g of crushed ice and the contents of the beaker were stirred well. A light yellow coloured solid that formed was filtered and washed with water (10 ml × 3). The crude solid was re-crystallized from EtOH to furnish 3.10 g of 1,1-di(methylsulfanyl)-3-(aryl)-1-propen-3-one in 80–92% yield as light yellow colored crystals.
3,3-Bis(methylthio)-1-phenylprop-2-en-1-one34 (1a). Pale yellow solid; yield 88%, mp. 94–96 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.48 (s, 3H), 2.64 (s, 3H), 6.86 (s, 1H), 7.49 (d, J = 7.6 Hz, 2H), 7.56 (d, J = 7.6 Hz, 1H), 7.94 (d, J = 6.8 Hz, 1H), 7.94 (m, 2H). 13C NMR (75 MHz, CDCl3) δC: 14.9, 17.2, 109.3, 127.6, 128.3, 131.6, 139.2, 166.3, 185.5. LC-MS calcd m/z 224, found 225 [(M + 1)]+.
3,3-Bis(methylthio)-1-(naphthalen-2-yl)prop-2-en-1-one34(1b). Yellow solid; yield 92%, mp. 96–98 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.49 (s, 3H), 2.71 (s, 3H), 7.05 (s, 1H), 7.57–7.64 (m, 2H), 7.95–8.02 (m, 3H), 8.10 (d, J = 7.2 Hz, 1H), 8.62 (s, 1H). 13C NMR (75 MHz, CDCl3) δC: 15.1, 17.4, 109.6, 124.3, 126.5, 127.6, 127.7, 128.4, 129.3, 132.6, 134.9, 136.6, 166.3, 185.5. LC-MS calcd m/z 274, found 275 [(M + 1)]+.
3,3-Bis(methylthio)-1-(naphthalen-1-yl)prop-2-en-1-one34 (1c). Yellow solid; yield 85%, mp. 79–82 °C; 1H NMR (300 MHz, CDCl3) δH: 2.46 (s, 3H), 2.57 (s, 3H), 6.56 (s, 1H), 7.45–7.54 (m, 2H), 7.70 (d, J = 8 Hz, 1H), 7.92 (d, J = 12 Hz, 1H). 13C NMR (75 MHz, CDCl3) δC: 14.8, 17.1, 113.5, 124.5, 125.8, 126.0, 126.8, 128.1, 130.1, 130.7, 133.6, 138.9, 165.9, 189.3. LC-MS calcd m/z 274, found 275 [(M + 1)]+.
1-(4-Chlorophenyl)-3,3-bis(methylthio)prop-2-en-1-one34 (1d). Yellow solid; yield 92%, mp. 104–106 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.47 (s, 3H), 2.65 (s, 3H), 6.84 (s, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.97 (d, J = 8.4 Hz, 2H). 13C NMR (75 MHz, CDCl3) δC: 14.9, 17.3, 108.9, 128.6, 129.0, 137.7, 183.8.* LC-MS calcd m/z 258, found 259 [(M + 1)]+. [* – Two carbon signals have merged together.]
3,3-Bis(methylthio)-1-p-tolylprop-2-en-1-one34 (1e). Yellow solid; yield 82%, mp. 98–100 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.49 (s, 3H), 2.63 (s, 3H), 2.78 (s, 1H), 6.84 (s, 1H), 7.23 (d, J = 8.8 Hz, 2H), 7.64 (d, J = 8.8 Hz, 2H). 13C NMR (75 MHz, CDCl3) δC: 15.0, 17.3, 55.4, 21.5, 109.6, 127.8, 129.1, 136.7, 142.3, 165.5, 185.4. LC-MS calcd m/z 238, found 239 [(M + 1)]+.
3,3-Bis(methylthio)-1-ferrocenyl-2-propen-1-one34 (1f). Yellow solid; yield: 85%, mp. 112–114 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.42 (s, 3H), 2.60 (s, 3H), 4.16 (s, 4H), 4.50 (s, 1H), 4.83 (s, 2H), 6.41 (s, 1H). 13C NMR (75 MHz, CDCl3) δC: 14.9, 17.4, 69.0, 69.8, 71.6, 81.6, 111.3, 160.9, 189.3. LC-MS calcd m/z 332, found 333 [(M + 1)]+.
3,3-Bis(methylthio)-1-pyrenyl-2-propen-1-one33 (1g). Yellow solid; yield: 80% yield, mp. 150–152 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.56 (s, 3H), 2.61 (s, 3H), 6.82 (s, 1H), 7.35 (s, 1H), 8.13 (t, J = 8 Hz, 1H), 8.22–8.29 (m, 3H), 8.33–8.38 (m, 4H), 8.63 (d, J = 9.2 Hz, 1H). 13C NMR (100 MHz, CDCl3) δC: 15.0, 17.3, 114.2, 124.2, 124.8, 124.9, 125.5, 126.2, 127.1, 128.3, 128.5, 128.7, 128.8, 130.6, 132.7, 135.7, 189.6.* LC-MS calcd m/z 348, found 349 [(M + 1)]+. [* – Two carbon signals have merged together.]
3,3-Bis(methylthio)-1-(3-nitrophenyl)prop-2-en-1-one38 (1h). Yellow solid; yield: 80%, mp. 110–112 °C; 1H NMR (400 MHz, CDCl3) δH: 2.57 (s, 3H), 2.62 (s, 3H), 6.74 (s, 1H), 7.64 (t, J = 8 Hz, 1H), 8.26 (d, J = 8 Hz, 1H), 8.35 (d, J = 8 Hz, 1H), 8.71 (s, 1H). 13C NMR (75 MHz, CDCl3) δC: 14.5, 16.8, 107.3, 121.7, 125.3, 128.9, 132.9, 140.1, 147.6, 169.6, 181.9. LC-MS calcd m/z 269, found 270 [(M + 1)]+.
1-(3-Methoxyphenyl)-3,3-bis(methylthio)prop-2-en-1-one38 (1i). Yellow solid; yield 84%, mp. 88–90 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.49 (s, 3H), 2.65 (s, 3H), 3.80 (s, 3H), 6.83 (s, 1H), 7.13 (d, J = 8 Hz, 1H), 7.41 (t, J = 8 Hz, 2H), 7.54 (d, J = 8 Hz, 1H). 13C NMR (75 MHz, CDCl3) δC: 14.9, 17.3, 55.3, 109.4, 112.4, 117.9, 119.9, 129.3, 140.7, 159.7, 166.5, 185.2. LC-MS calcd m/z 254, found 255 [(M + 1)]+.
1-(2-Fluorophenyl)-3,3-bis(methylthio)prop-2-en-1-one38 (1j). Yellow solid; yield: 84%, mp. 70–72 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.50 (s, 3H), 2.58 (s, 3H), 6.66 (s, 1H), 7.28–7.33 (m, 2H), 7.55–7.61 (m, 1H), 7.75 (t, J = 8 Hz, 1H). 13C NMR (75 MHz, CDCl3) δC: 15.1, 17.3, 113.2, 113.3, 116.0, 116.3, 124.4, 127.5, 127.7, 131.2, 132.9, 133.1, 158.8, 162.1, 167.0, 182.3. LC-MS calcd m/z 242, found 243 [(M + 1)]+.
3,3-Bis(methylthio)-1-(3-(trifluoromethyl)phenyl)prop-2-en-1-one38 (1k). Yellow solid; yield: 85%, mp. 88–90 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.49 (s, 3H), 2.68 (s, 3H), 6.90 (s, 1H), 7.74 (t, J = 8 Hz, 1H), 7.92 (d, J = 8 Hz, 1H), 8.18 (s, 1H), 8.28 (d, J = 7.6 Hz, 1H). 13C NMR (75 MHz, CDCl3) δC: 15.0, 17.3, 108.4, 124.4, 128.0, 128.9, 130.7, 131.1, 139.9, 168.8, 183.7. LC-MS calcd m/z 292, found 293 [(M + 1)]+.
1-(4-Bromophenyl)-3,3-bis(methylthio)prop-2-en-1-one39 (1l). Yellow solid; yield: 80%, mp. 100–104 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.50 (s, 3H), 2.66 (s, 3H), 6.84 (s, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H). 13C NMR (75 MHz, CDCl3) δC: 14.4, 16.8, 108.3, 125.8, 129.0, 131.1, 137.5, 167.1, 183.2.* LC-MS calcd m/z 303, found 304 [(M + 1)]+. [* – Two carbon signals have merged together.]
1-(4-Methoxyphenyl)-3,3-bis(methylthio)prop-2-en-1-one39 (1m). Yellow solid; yield: 80%, mp. 100–102 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.45 (s, 3H), 2.63 (s, 3H), 3.82 (s, 1H), 6.84 (s, 1H), 7.00 (d, J = 8.8 Hz, 2H), 7.94 (d, J = 8.8 Hz, 2H). 13C NMR (75 MHz, CDCl3) δC: 15.1, 17.3, 55.4, 109.8, 113.7, 129.4, 132.2, 162.6, 164.6, 184.6.* LC-MS calcd m/z 254, found 255 [(M + 1)]+. [* – Two carbon signals have merged together.]
1-(2,4-Dichlorophenyl)-3,3-bis(methylthio)prop-2-en-1-one40 (1n). Yellow solid; yield: 82%, mp. 108–110 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.49 (s, 3H), 2.54 (s, 3H), 6.44 (s, 1H), 7.50 (d, J = 9.2 Hz, 1H), 7.58 (d, J = 8 Hz, 1H), 7.68 (s, 1H). 13C NMR (75 MHz, CDCl3) δC: 15.0, 17.2, 112.5, 127.2, 129.9, 130.8, 131.7, 136.2, 138.8, 167.7, 184.9. LC-MS calcd m/z 293, found 294 [(M + 1)]+.
1-(3,4-Difluorophenyl)-3,3-bis(methylthio)prop-2-en-1-one (1o). Yellow solid; yield: 83%, mp. 137 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.49 (s, 3H), 2.68 (s, 3H), 6.81 (s, 1H), 7.43–7.48 (m, 1H), 7.65 (d, J = 6.8 Hz, 1H). 13C NMR (75 MHz, CDCl3) δC: 15.0, 17.3, 106.7, 108.1, 110.3, 110.7, 142.5, 161.1, 161.3, 164.4, 164.6, 169.5, 182.3. LC-MS calcd m/z 260 found 261 [(M + 1)]+. HRMS (ESI-TOF) calcd for C11H10F2OS2Na [M + Na]+ 283.0039 found 283.0034.
1-(3,4-Dichlorophenyl)-3,3-bis(methylthio)prop-2-en-1-one (1p). Yellow solid; yield: 79%, mp. 118–120 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.48 (s, 3H), 2.67 (s, 3H), 6.83 (s, 1H), 7. 73 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H), 8.14 (s, 1H). 13C NMR (75 MHz, CDCl3) δC: 15.1, 17.4, 108.1, 126.7, 129.6, 130.4, 132.8, 135.9, 138.9, 168.9, 182.6. LC-MS calcd m/z 293, found 294 [(M + 1)]+. HRMS (ESI-TOF) calcd for C11H10Cl2OS2Na [M + Na]+ 314.9448 found 314.9440.
1-(3-Bromophenyl)-3,3-bis(methylthio)prop-2-en-1-one (1q). Yellow solid; yield: 84%, mp. 78–80 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.48 (s, 3H), 2.67 (s, 3H), 6.83 (s, 1H), 7.46 (t, J = 8 Hz, 1H), 7.75 (d, J = 8 Hz, 1H), 7.96 (d, J = 7.6 Hz, 1H), 8.07 (s, 1H). 13C NMR (75 MHz, CDCl3) δC: 15.1, 17.5, 108.7, 122.7, 126.3, 130.1, 130.8, 134.5, 141.3, 168.3, 183.9. LC-MS calcd m/z 303, found 304 [(M + 1)]+. HRMS (ESI-TOF) calcd for C11H11BrOS2Na [M + Na]+ 324.9332 found 324.9326.
3,3-Bis(methylthio)-1-(2-(trifluoromethyl)phenyl)prop-2-en-1-one (1r). Pale yellow solid; yield: 84%, mp. 108–110 °C; 1H NMR (300 MHz, CDCl3) δH: 2.46 (s, 3H), 2.54 (s, 3H), 6.26 (s, 1H), 7.27–7.61 (m, 3H), 7.69 (d, J = 8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δC: 16.9, 19.1, 78.6, 79.0, 79.4, 114.1, 128.2, 128.3, 129.1, 130.1, 131.3, 133.7, 143.4, 169.8, 189.5. LC-MS calcd m/z 292, found 293 [(M + 1)]+. HRMS (ESI-TOF) calcd for C12H11F3OS2Na [M + Na]+ 315.0101 found 315.0093.
1-(2-Fluoro-5-(trifluoromethyl)phenyl)-3,3-bis(methylthio)prop-2-en-1-one (1s). Yellow solid; yield: 70%, mp. 88–90 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.51 (s, 3H), 2.58 (s, 3H), 6.66 (s, 1H), 7.57 (t, J = 9.2 Hz, 1H), 7.95–8.02 (m, 1H). 13C NMR (75 MHz, CDCl3) δC: 15.3, 17.4, 112.4, 117.1, 119.4, 124.8, 126.3, 127.5, 128.2, 129.2, 129.9, 160.8, 163.3, 169.6, 180.4. LC-MS calcd m/z 310, found 311 [(M + 1)]+. HRMS (ESI-TOF) calcd for C12H10F4OS2Na [M + Na]+ 333.0007 found 333.0003.
1-(4-Iodophenyl)-3,3-bis(methylthio)prop-2-en-1-one (1t). Yellow solid; yield: 77%, mp. 88–90 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.49 (s, 3H), 2.64 (s, 3H), 6.82 (s, 1H), 7.73 (d, J = 8 Hz, 1H), 7.86 (d, J = 8 Hz, 1H). 13C NMR (75 MHz, CDCl3) δC: 15.1, 17.4, 99.1, 108.7, 128.0, 129.3, 137.7, 138.7, 167.7, 184.68.* LC-MS calcd m/z 350, found 351 [(M + 1)]+. HRMS (ESI-TOF) calcd for C11H11IOS2Na [M + Na]+ 372.9194 found 372.9190. [* – Two carbon signals have merged together.]
1-(2-Fluoro-4-(trifluoromethyl)phenyl)-3,3-bis(methylthio)prop-2-en-1-one (1u). Yellow solid; yield: 72%, mp. 84–86 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.51 (s, 3H), 2.57 (s, 3H), 6.63 (s, 1H), 7.67 (d, J = 8 Hz, 1H), 7.79 (d, J = 10.4 Hz, 1H), 7.92 (t, J = 8 Hz, 1H). 13C NMR (75 MHz, CDCl3) δC: 15.2, 17.4, 112.4, 113.8, 113.9, 114.0, 121.3, 121.4, 121.5, 124.3, 130.7, 132.2, 134.3, 134.7, 158.6, 161.2, 169.6, 180.7. LC-MS calcd m/z 310, found 311 [(M + 1)]+. HRMS (ESI-TOF) calcd for C12H10F4OS2Na [M + Na]+ 333.0007 found 333.0001.
1-(4-Chloro-3-(trifluoromethyl)phenyl)-3,3-bis(methylthio)prop-2-en-1-one (1v). Yellow solid; yield: 77%, mp. 126–128 °C; 1H NMR (400 MHz, DMSO-d6) δH: 2.49 (s, 3H), 2.68 (s, 3H), 6.88 (s, 1H), 7.85 (d, J = 8 Hz, 1H), 8.25 (s, 1H), 8.29 (d, J = 8 Hz, 1H). 13C NMR (75 MHz, CDCl3) δC: 15.2, 17.5, 107.9, 121.2, 123.9, 126.8, 126.9, 128.5, 128.8, 131.7, 135.0, 135.6, 137.9, 169.8, 182.7. LC-MS calcd m/z 326, found 327 [(M + 1)]+. HRMS (ESI-TOF) calcd for C12H10ClF3OS2Na [M + Na]+ 348.9711 found 348.9708.
Method I (conventional heating method). To the mixture of AKDTA 1 (1 mmol), o-phenylenediamine 2 (1 mmol), acetic acid (40 mol%) in water (6 ml) was added and heated at 100 °C for 2 h. The reaction mixture was treated with sodium bicarbonate and extracted with ethyl acetate. The combined ethyl acetate extracts were washed with water, dried and concentrated under rotary vacuum evaporation. The crude residue was recrystallized to obtain pure solid product 4a–v (80–95% yields).
Method II (microwave irradiation method). A 10 ml glass vial sealed by a septum, containing a mixture of AKDTA 1 (1 mmol) o-phenylenediamine 2 (1 mmol), and acetic acid (40 mol%) in water (6 ml) was placed in a microwave synthesizer. The vial was then subjected to microwave irradiation programmed at 120 W, 100 °C with 1 bar pressure. After completion of the reaction (5 min), the vial was cooled to room temperature and extracted with ethyl acetate and the crude was purified by recrystallization in ethanol to yield pure 4a–v (78–90%).
2-Phenyl-1H-benzo[d]imidazole41 (4a). Off white crystalline solid; yield: 95% (thermal), 90% (MW), mp. 296 °C; UV λmax (MeOH) = 241 nm (log ε = 2.39), 202 nm (log ε = 2.61). 1H NMR (400 MHz, DMSO-d6) δH: 7.15–7.22 (m, 2H), 7.46–7.75 (m, 4H), 7.65 (d, J = 7.6 Hz, 1H), 8.16 (d, J = 6.8 Hz, 2H), 12.86 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 111.3, 118.9, 121.7, 122.5, 126.4, 128.9, 129.8, 130.2, 135.0, 143.8, 151.2. LC-MS calcd m/z: 194, found 195 [(M + 1)]+.
2-(Naphthalene-2-yl)-1H-benzo[d]imidazole42 (4b). Off white solid; yield: 92% (thermal), 90% (MW), mp. 214–215 °C; UV λmax (MeOH) = 316 nm (log ε = 2.75), 281 nm (log ε = 2.65), 241 nm (log ε = 2.88). 1H NMR (400 MHz, DMSO-d6) δH: 7.19–7.23 (m, 2H), 7.54–7.69 (m, 4H), 7.97–8.08 (m, 3H), 8.30 (d, J = 8.4 Hz, 1H), 8.72 (s, 1H), 13.03 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 129.0, 131.1, 131.6, 131.8, 132.7, 133.3, 133.3, 137.9, 138.6, 156.7.* LC-MS calcd m/z: 244 found 245 [(M + 1)]+. [* – Two carbon signals have merged together.]
2-(Naphthalene-1-yl)-1H-benzo[d]imidazole41 (4c). Off white solid; yield: 93% (thermal), 87% (MW), mp 270–272 °C; UV λmax (MeOH) = 306 nm (log ε = 2.58), 226 nm (log ε = 2.88). 1H NMR (400 MHz, DMSO-d6) δH: 7.22–7.26 (m, 2H), 7.58–7.68 (m, 5H), 7.80–8.04 (m, 2H), 8.08 (d, J = 8.4 Hz, 1H), 9.10 (d, J = 7.6 Hz, 1H), 12.92 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 122.4, 125.5, 126.6, 126.8, 127.3, 128.3, 128.7, 130.4, 131.1, 134.0, 151.8. LC-MS calcd m/z: 244, found 245 [(M + 1)]+.
2-(4-Chlorophenyl)-1H-benzo[d]imidazole41 (4d). Off white solid; yield: 88% (thermal), 82% (MW), mp. 290–292 °C; UV (MeOH) λmax = 307 nm (log ε = 2.77), 245 nm (log ε = 2.45). 1H NMR (400 MHz, DMSO-d6) δH: 7.20 (s, 2H), 7.52 (d, J = 6.8 Hz, 1H), 7.61 (d, J = 8.8 Hz, 2H), 7.66 (s, 1H), 8.17 (d, J = 8.4 Hz, 2H), 12.94 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 122.5, 128.3, 129.1, 129.3, 135.3, 150.7.* LC-MS calcd m/z: 228, found 229 [(M + 1)]+. [* – Two carbon signals have merged together.]
2-p-Tolyl-1H-benzo[d]imidazole41 (4e). Off white solid; yield: 87% (thermal), 82% (MW), mp. 275–277 °C; UV λmax (MeOH) = 304 nm (log ε = 2.77), 243 nm (log ε = 2.53). 1H NMR (400 MHz, DMSO-d6) δH: 7.13–7.20 (m, 2H), 7.34 (d, J = 8 Hz, 2H), 7.49 (d, J = 6.4 Hz, 1H), 7.62 (d, J = 7.6 Hz, 1H), 8.05 (d, J = 8 Hz, 2H), 12.76 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 22.0, 112.8, 127.4, 128.4, 130.3, 140.4, 152.5.* LC-MS calcd m/z: 208, found 209 [(M + 1)]+. [* – Two carbon signals have merged together.]
2-(Ferrocenyl-2-yl)-1H-benzo[d]imidazole43 (4f). Off white solid; yield: 88% (thermal), 83% (MW), mp. 300 °C; UV λmax (MeOH) = 304 nm (log ε = 2.70), 206 nm (log ε = 2.93). 1H NMR (400 MHz, DMSO-d6) δH: 4.08 (s, 4H), 4.45 (s, 2H), 5.02 (s, 2H), 7.08–7.15 (m, 2H), 7.42 (d, J = 6.8 Hz, 1H), 7.52 (d, J = 7.6 Hz, 1H), 12.30 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 60.7, 69.8, 70.2, 74.8, 110.9, 118.4, 121.8, 153.4. LC-MS calcd m/z: 303, found 304 [(M + 1)]+.
2-(Pyren-2-yl)-1H-benzo[d]imidazole (4g). Off white solid; yield: 90% (thermal), 87% (MW), mp. 300 °C; UV λmax (MeOH) = 351 nm (log ε = 3.51), 278 nm (log ε = 2.69), 241 nm (log ε = 2.87). IR (KBr): 3423, 3043, 1743, 1649, 1421, 1278, 1018, 842, 746 cm−1. 1H NMR (400 MHz, DMSO-d6) δH: 7.26–7.28 (m, 2H), 7.71–7.74 (m, 2H), 8.20 (t, J = 7.6 Hz, 1H), 8.27–8.37 (m, 3H), 8.45 (d, J = 8 Hz, 1H), 8.56 (d, J = 8 Hz, 1H), 9.51 (d, J = 9.2 Hz, 1H), 13.13 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 122.6, 124.2, 124.8, 125.1, 125. 3, 126.0, 126.1, 126.4, 127.1, 127.8, 128.9, 129.1, 130.8, 131.4, 132.0. LC-MS calcd m/z: 318, found 319 [(M + 1)]+. HRMS (ESI-TOF) calcd for C23H16N2Na [M + Na]+ 343.1211 found 343.1208.
2-(3-Nitrophenyl)-1H-benzo[d]imidazole44 (4h). Off white solid; yield: 89% (thermal), 82% (MW), mp. 208–210 °C; UV λmax (MeOH) = 306 nm (log ε = 2.64), 245 nm (log ε = 2.52). 1H NMR (400 MHz, DMSO-d6) δH: 7.20–7.28 (m, 2H), 7.57 (d, J = 7.6 Hz, 1H), 7.71 (d, J = 8 Hz, 1H), 7.85 (t, J = 8 Hz, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.60 (d, J = 8 Hz, 1H), 9.00 (s, 1H), 13.25 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 111.8, 119.5, 121.3, 122.3, 123.4, 124.1, 130.4, 132.2, 132.7, 135.4, 143.9, 148.6, 149.4. LC-MS calcd m/z: 239, found 240 [(M + 1)]+.
2-(3-Methoxyphenyl)-1H-benzo[d]imidazole45 (4i). Off white solid; yield: 88% (thermal), 83% (MW), mp. 210–212 °C; UV λmax (MeOH) = 306 nm (log ε = 2.74), 210 nm (log ε = 2.91). 1H NMR (400 MHz, DMSO-d6) δH: 3.85 (s, 3H), 7.03–7.06 (m, 1H), 7.19 (s, 2H), 7.44 (t, J = 8.4 Hz, 1H), 7.53 (s, 1H), 7.64 (s, 1H), 7.73–7.75 (m, 2H), 12.84 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 55.8, 111.9, 116.3, 119.2, 122.6, 130.6, 131.9, 151.6, 160.1.* LC-MS calcd m/z: 224, found 225 [(M + 1)]+. [* – Two carbon signals have merged together.]
2-(2-Fluorophenyl)-1H-benzo[d]imidazole (4j). Off white solid; yield: 82% (thermal), 79% (MW), mp. 205–206 °C; UV λmax (MeOH) = 303 nm (log ε = 2.87), 206 nm (log ε = 3.02). IR (KBr): 3425, 2854, 1739, 1423, 1018, 748 cm−1. 1H NMR (400 MHz, DMSO-d6) δH: 7.12–7.25 (m, 2H), 7.36–7.45 (m, 2H), 7.52–7.58 (m, 2H), 7.68 (d, J = 7.2 Hz, 1H), 8.23 (t, J = 8 Hz, 1H), 12.53 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 115.9, 116.3, 117.3, 117.5, 125.1, 125.1, 130.1, 130.1, 131.5, 131.6, 131.6, 146.9, 158.6, 161.9. LC-MS calcd m/z: 212, found 213 [(M + 1)]+ HRMS (ESI-TOF) calcd for C13H9FN2Na [M + Na]+ 235.0647 found 235.0641.
2-(3-(Trifluoromethyl)phenyl)-1H-benzo[d]imidazole46 (4k). Off white solid; yield: 84% (thermal), 80% (MW), mp. 206–208 °C; UV λmax (MeOH) = 306 nm (log ε = 2.77), 206 nm (log ε = 2.90). 1H NMR (400 MHz, DMSO-d6) δH: 7.19–7.26 (m, 2H), 7.56 (d, J = 7.6 Hz, 1H), 7.69 (d, J = 7.2 Hz, 1H) 7.77–7.85 (m, 2H), 8.46 (d, J = 7.6 Hz, 1H), 8.51 (s, 1H), 13.12 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 111.4, 119.3, 122.2, 123.1, 123.6, 126.0, 129.4, 130.0, 130.7, 131.1, 131.4, 135.2, 150.3. LC-MS calcd m/z: 262, found 263 [(M + 1)]+.
2-(4-Bromophenyl)-1H-benzo[d]imidazole44 (4l). Off white solid; yield: 90% (thermal), 85% (MW), mp. 296–298 °C; UV λmax (MeOH) 308 nm (log ε = 2.72), 246 nm (log ε = 2.46). 1H NMR (400 MHz, DMSO-d6) δH: 7.16–7.24 (m, 2H), 7.52 (d, J = 7.2 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.75 (d, J = 8.4 Hz, 2H), 8.10 (d, J = 8.4 Hz, 2H), 12.94 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 122.6, 123.8, 128.6, 129.7, 132.0, 150.8.* LC-MS calcd m/z: 212, found 213 [(M + 1)]+. [* – Two carbon signals have merged together.]
2-(4-Methoxyphenyl)-1H-benzo[d]imidazole44 (4m). Off white solid; yield: 90% (thermal), 88% (MW), mp. 225–226 °C; UV λmax (MeOH) = 307 nm (log ε = 2.82), 248 nm (log ε = 2.52). 1H NMR (400 MHz, DMSO-d6) δH: 3.82 (s, 3H), 7.09 (d, J = 8.8 Hz, 2H), 7.14–7.15 (m, 2H), 7.46–7.59 (m, 2H), 8.09 (d, J = 8.8 Hz, 2H), 12.68 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 14.6, 122.1, 123.2, 128.4, 151.9, 161.0.* LC-MS calcd m/z: 224, found 225 [(M + 1)]+. [* – Two carbon signals have merged together.]
2-(2,4-Dichlorophenyl)-1H-benzo[d]imidazole47 (4n). Off white solid; yield: 85% (thermal), 81% (MW), mp. 216–218 °C; UV λmax (MeOH) = 296 nm (log ε = 2.56), 207 nm (log ε = 2.95). 1H NMR (400 MHz, DMSO-d6) δH: 7.23–7.24 (m, 2H), 7.59–7.62 (m, 3H), 7.82 (s, 1H), 7.93 (d, J = 8.4 Hz, 1H), 12.72 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 127.4, 132.2, 133.5, 134.3, 137.6, 137.8, 140.5, 153.2.* LC-MS calcd m/z: 262, found 263 [(M + 1)]+. [* – Two carbon signals have merged together.]
2-(3,4-Difluorophenyl)-1H-benzo[d]imidazole (4o). Off white solid; yield: 84% (thermal), 78% (MW), mp. 230 °C; UV λmax (MeOH) = 306 nm (log ε = 2.69), 242 nm (log ε = 2.39). IR (KBr): 3445, 3300, 1620, 1400, 1200, 1080, 760 cm−1. 1H NMR (400 MHz, DMSO-d6) δH: 7.19–7.28 (m, 2H), 7.36–7.40 (m, 1H), 7.55 (d, J = 8 Hz, 1H), 7.68 (d, J = 7.6 Hz, 1H), 7.84 (d, J = 7.4 Hz, 2H), 13.05 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 104.9, 105.2, 105.6, 109.6, 109.9, 111.9, 119.6, 122.5, 123.5, 133.8, 133.9, 134.1, 135.3, 143.9, 149.3, 161.4, 161.6, 164.7, 164.88. LC-MS calcd m/z: 230, found 231 [(M + 1)]+. HRMS (ESI-TOF) calcd for C13H8F2N2Na [M + Na]+ 253.0553 found 253.0548.
2-(3,4-Dichlorophenyl)-1H-benzo[d]imidazole (4p). Off white crystalline solid; yield: 83% (thermal), 80% (MW), mp. 235 °C; UV λmax (MeOH) = 294 nm (log ε = 2.60), 205 nm (log ε = 2.90). IR (KBr): 3439, 3380, 1590, 1590, 1312, 1020, 800 cm−1. 1H NMR (400 MHz, DMSO-d6) δH: 7.23 (t, J = 8.4 Hz, 2H), 7.54 (d, J = 7.2 Hz, 1H), 7.67 (d, J = 7.6 Hz, 1H), 7.83 (d, J = 8.4 Hz, 1H), 8.14 (d, J = 8.8 Hz, 1H), 8.38 (s, 1H), 13.05 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 115.8, 122.7, 127.8, 129.2, 130.2, 132.9, 133.5, 135.5, 139.3, 148.5.* LC-MS calcd m/z: 263, found 264 [(M + 1)]+. HRMS (ESI-TOF) calcd for C13H8Cl2N2Na [M + Na]+ 284.9962 found 284.9955. [* – Two carbon signals have merged together.]
2-(3-Bromophenyl)-1H-benzo[d]imidazole44 (4q). Off white solid; yield: 89% (thermal), 84% (MW), mp. 262–264 °C; UV λmax (MeOH) = 306 nm (log ε = 2.79), 207 nm (log ε = 2.95). 1H NMR (400 MHz, DMSO-d6) δH: 7.19–7.27 (m, 2H), 7.50–7.56 (m, 2H), 7.67–7.70 (m, 2H), 8.18 (d, J = 8 Hz, 1H), 8.37 (s, 1H), 13.00 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 155.0, 137.4, 137.2, 135.4, 130.2, 127.5.* LC-MS calcd m/z: 273, found 274 [(M + 1)]+. [* – Two carbon signals have merged together.]
2-(2-(Trifluoromethyl)phenyl)-1H-benzo[d]imidazole48 (4r). White crystalline solid; yield: 83% (thermal), 79% (MW), mp. 274–276 °C; UV λmax (MeOH) = 282 nm (log ε = 2.47), 206 nm (log ε = 2.92). 1H NMR (400 MHz, DMSO-d6) δH: 7.18–7.26 (m, 2H), 7.52 (d, J = 8 Hz, 1H), 7.67 (d, J = 7.6 Hz, 1H), 7.74–7.85 (m, 3H), 7.93 (d, J = 7.6 Hz, 1H), 12.72 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 127.1, 131.1, 133.5, 134.5, 136.5, 137.1, 154.4.* LC-MS calcd m/z: 262, found 263 [(M + 1)]+. [* – Two carbon signals have merged together.]
2-(2-Fluoro-5-(trifluoromethyl)phenyl)-1H-benzo[d]imidazole (4s). Off white crystalline solid; yield: 80% (thermal), 78% (MW), mp. 215 °C; UV λmax (MeOH) = 307 nm (log ε = 2.73), 206 nm (log ε = 2.89). IR (KBr): 3441, 3053, 2924, 1789, 1404, 1332, 1159, 752 cm−1. 1H NMR (400 MHz, DMSO-d6) δH: 7.24–7.30 (m, 2H), 7.61 (d, J = 7.6 Hz), 7.75 (dd, J = 8.4 Hz, J = 7.2 Hz, 2H), 7.94 (d, J = 10.8 Hz, 1H), 8.46 (t, J = 8 Hz, 1H), 12.77 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 105.2, 109.6, 109.9, 111.9, 119.6, 122.5, 123.5, 133.9, 134.1, 135.3, 143.9, 149.3, 161.6, 164.7. LC-MS calcd m/z: 280, found 281 [(M + 1)]+. HRMS (ESI-TOF) calcd for C14H8F4N2Na [M + Na]+ 303.0521 found 303.0516.
2-(4-Iodophenyl)-1H-benzo[d]imidazole49 (4t). Off white crystalline solid; yield: 90% (thermal), 86% (MW), mp. 290–292 °C; UV λmax (MeOH) = 310 nm (log ε = 2.82), 252 nm (log ε = 2.53). 1H NMR (400 MHz, DMSO-d6) δH: 7.16–7.19 (m, 1H), 7.56–7.58 (m, 1H), 7.92 (dd, J = 8 Hz, 8.4 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δC: 96.2, 122.4, 128.6, 130.2, 137.9, 150.9.* LC-MS calcd m/z: 320, found 321 [(M + 1)]+. [* – Two carbon signals have merged together.]
2-(2-Fluoro-4-(trifluoromethyl)phenyl)-1H-benzo[d]imidazole (4u). Off white solid; yield: 87% (thermal), 82% (MW), mp. 230 °C; UV λmax (MeOH) = 308 nm (log ε = 2.75), 206 nm (log ε = 2.85). IR (KBr): 3441, 3086, 1770, 1444, 1332, 1130, 740 cm−1. 1H NMR (400 MHz, DMSO-d6) δH: 7.24–7.30 (m, 2H), 7.61 (d, J = 7.6 Hz, 1H), 7.75 (dd, J = 8.4 Hz, J = 7.2 Hz, 2H), 7.94 (d, J = 10.8 Hz, 1H), 8.46 (t, J = 8 Hz, 1H), 12.77 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 112.4, 113.8, 114.1, 119.4, 121.7, 122.4, 123.5, 131.4, 135.4, 143.2, 157.7, 161.1. LC-MS calcd m/z: 280, found 281 [(M + 1)]+. HRMS (ESI-TOF) calcd for C14H8F4N2Na [M + Na]+ 303.0521 found 303.0516.
2-(4-Chloro-3-(trifluoromethyl)phenyl)-1H-benzo[d]imidazole (4v). Off white solid; yield: 87% (thermal), 83% (MW), mp. 200 °C; UV λmax (MeOH) = 309 nm (log ε = 2.78), 246 nm (log ε = 2.50). IR (KBr): 3444, 3053, 1618, 1438, 1317, 1180, 743 cm−1. 1H NMR (400 MHz, DMSO-d6) δH: 7.20–7.28 (m, 2H), 7.56 (d, J = 7.6 Hz, 1H), 7.69 (d, J = 8 Hz, 1H), 7.92 (d, J = 8.4 Hz, 1H), 8.44 (d, J = 8.4 Hz, 1H), 8.61 (s, 1H), 13.18 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC: 129.3, 130.6, 133.1, 133.5, 134.4, 135.7, 136.6, 137.6, 154.2.* LC-MS calcd m/z: 296, found 297 [(M + 1)]+. HRMS (ESI-TOF) calcd for C14H8ClF3N2Na [M + Na]+ 319.0226 found 319.0220. [* – Two carbon signals have merged together.]
Acknowledgements
SS thanks DST-MRP, New Delhi and UGC – MRP, New Delhi for the financial assistances. PD thanks UGC for meritorious fellowship. We thank DST-IRHPA for fundings towards higher resolution NMR spectrometer. We thank Prof. H. Surya Prakash Rao, Department of Chemistry, Pondicherry University, Puducherry for generous help in recording spectra and helpful discussions. SS dedicates this manuscript to his mentor Prof. H. Surya Prakash Rao, Department of Chemistry, Pondicherrry University, Puducherry-14.Notes and references
- A. W. White, N. J. Curtin, B. W. Eastman, B. T. Golding, Z. Hostomsky, S. Kyle, K. A. Maegley, D. J. Skalitzki, S. E. Webber, X.-H. Yu and R. J. Griffin, Bioorg. Med. Chem. Lett., 2004, 14, 2433–2437 CrossRef CAS PubMed.
- A. Lee- Dutra, K. L. Arienti, D. J. Buzard, M. D. Hack, H. Khatuya, P. J. Desai, S. Nguyen, R. L.Thurmond, L. Karlsson, J. P. Edwards and J. G. Breitenbucher, Bioorg. Med. Chem. Lett., 2006, 16, 6043–6048 CrossRef CAS PubMed.
- R. C. Rivera and O. Munoz, J. Med. Microbiol., 1992, 37, 221–224 CrossRef PubMed.
- T. Güngör, A. Fouquet, J.-M. Teulon, D. Provost, M. Cazes and A. Cloarec, J. Med. Chem., 1992, 35, 4455–4563 CrossRef.
- W. A. Denny, G. W. Rewcastle and B. C. Baguley, J. Med. Chem., 1990, 33, 814–819 CrossRef CAS.
- E. Seyhan, N. Sultan, A. Nilgun and N. Noyanalpan, Arzneim. Forsch., 1997, 47, 410–412 Search PubMed.
- P. C. Santhosh, S. N. Pandeya and K. P. Ashish, Int. J. Res. Ayurveda Pharm., 2011, 6, 1726–1737 Search PubMed.
- (a) R. K. Dieter, Tetrahedron, 1986, 42, 3029–3096 CrossRef CAS; (b) H. Junjappa, H. Ila and C. V. Ashokan, Tetrahedron, 1990, 46, 5423–5506 CrossRef CAS.
- A. R. Katritzky and A. J. Boulton, Advances in Heterocyclic Chemistry, vol. 27, Academic, New York, 1980, p. 241 Search PubMed.
- S. P. Siva, M. Ritu and C. J. Subhash, Curr. Org. Chem., 2012, 16, 1905–1919 CrossRef.
- C. Anshul, K. Gurpreet and K. S. Anil, Int. J. Pharm. Phytopharm. Res., 2012, 3, 148–159 Search PubMed.
- P. N. Preston, A. Weissberger and E. C. Taylor, Chemistry of Heterocyclic Compounds; Part-1, Wiley, New York, 1981, vol. 40, pp. 6–60 Search PubMed.
- A. R. Kartritzky and C. W. Rees, Comprehensive Heterocyclic Chemistry, Pergamon, Oxford, 1984, vol. 5, pp. 457–474 Search PubMed.
- R. R. Nagawade and B. D. Shinde, Chin. Chem. Lett., 2006, 17, 453–456 CAS.
- H. Xiangming, M. Huiqiang and W. Yulu, ARKIVOC, 2007, 150–154 CrossRef.
- R. Trivedi, S. K. De and R. A. Gibbs, J. Mol. Catal. A: Chem., 2006, 245, 8–11 CrossRef CAS PubMed.
- P. Gogoi and D. Konwar, Tetrahedron Lett., 2006, 47, 79–82 CrossRef CAS PubMed.
- M. Chakrabarty, S. Karmakar, R. Mukherjee, S. Arima and Y. Harigaya, Monatsh. Chem., 2009, 140, 375–380 CrossRef CAS.
- C. Mukhopadhyay and P. K. Tapaswi, Tetrahedron Lett., 2008, 49, 6237–6240 CrossRef CAS PubMed.
- B. Das, H. Holla and Y. Srinivas, Tetrahedron Lett., 2007, 48, 61–64 CrossRef CAS PubMed.
- H. Sharghi, O. Asemani and R. Khalifeh, Synth. Commun., 2008, 38, 1128–1136 CrossRef CAS.
- T. Khan, T. Parvin and L. H. Choudhury, Synth. Commun., 2009, 40, 2339–2346 CrossRef.
- M. Kidwai and A. Jahan, J. Chem. Sci., 2010, 122, 607–612 CrossRef CAS PubMed.
- G. Renard and D. A. Lerner, New J. Chem., 2007, 31, 1417–1420 RSC.
- Y. X. Chen, L.-F. Qian, W. Zhang and B. Han, Angew. Chem., Int. Ed., 2008, 47, 9330–9333 CrossRef CAS PubMed.
- For recent reviews and references on benzimidazole synthesis: (a) L. C. R. Carvalho, E. Fernandes and M. M. B. Marques, Chem.–Eur. J., 2011, 17, 12544–12555 CrossRef CAS PubMed; (b) C. T. Brain and J. T. Steer, J. Org. Chem., 2003, 68, 6814–6816 CrossRef CAS PubMed; (c) K. Hirano, A. T. Biju and F. Glorius, J. Org. Chem., 2009, 74, 9570–9572 CrossRef CAS PubMed; (d) N. Zheng, K. W. Anderson, X. Huang, H. N. Nguyen and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 7509–7512 CrossRef CAS PubMed; (e) G. Brasche and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 1932–1934 CrossRef CAS PubMed; (f) R. K. Kumar and T. Punniyamurty, RSC Adv., 2012, 2, 4616–4619 RSC; (g) P. Saha, M. A. Ali, P. Ghosh and T. Punniyamurty, Org. Biomol. Chem., 2010, 8, 5692–5699 RSC; (h) H. Jin, X. Xu, J. Gao, J. Zhong and Y. Wang, Adv. Synth. Catal., 2010, 352, 347–350 CrossRef CAS; (i) S. Fu, H. Jiang, Y. Deng and W. Zeng, Adv. Synth. Catal., 2011, 353, 2795–2804 CrossRef CAS.
- Y. Kim, M. R. Kumar, N. Park, Y. Heo and S. Lee, J. Org. Chem., 2011, 76, 9577–9583 CrossRef CAS PubMed.
- L. Keurulainen, O. Salin, A. Siiskonen, J. M. Kern, P. Kiuru, M. Maass, J. Y. Kauhaluoma and P. Vuorela, J. Med. Chem., 2010, 53, 7664–7674 CrossRef CAS PubMed.
- Z. H. Zhang, L. Yin and Y. M. Wang, Catal. Commun., 2007, 8, 1126–1131 CrossRef CAS PubMed.
- S. Y. Lin, Y. Isome, E. Stewart, J. F. Liu, D. Yohanees and L. Yu, Tetrahedron Lett., 2006, 47, 2883–2886 CrossRef CAS PubMed.
- X. Diao, Y. Wang, Y. Jiang and D. Ma, J. Org. Chem., 2009, 74, 7974–7977 CrossRef CAS PubMed.
- H. Nishioka, Y. Ohmori, Y. Iba, E. Tsuda and T. Harayama, Heterocycles, 2004, 64, 193–198 CrossRef CAS PubMed.
- H. S. P. Rao and S. Sivakumar, Beilstein J. Org. Chem., 2007, 3, 31 CrossRef PubMed.
- H. S. P. Rao and S. Sivakumar, J. Org. Chem., 2006, 71, 8715–8723 CrossRef CAS PubMed.
- (a) L. O. Randall and B. Kappel, in Benzodiazepines, S. Garattini, E. Mussini and L. O. Randall, Raven Press, New York, 1973, p. 27 Search PubMed; (b) H. Schutz, Benzodiazepines, Springer, Heidel-berg, Germany, 1982 Search PubMed; (c) J. K. Landquist, in Comprehensive Heterocyclic Chemistry, A. R. Katritzky and C. W. Rees, Oxford, Pergamon, U.K., 1984, vol. 1, p. 166 Search PubMed; (d) G. A. Archer and L. H. Sternbach, Chem. Rev., 1968, 68, 747–784 CrossRef CAS; (e) R. Langnickel, R. Bluth and T. Ott, Pharmazie, 1986, 41, 689–694 CAS; (f) A. L. Parola, H. I. Yamamura and H. E. Laird, Life Sci., 1993, 52, 1329–1342 CrossRef CAS.
- S. Michelini, G. B. Cassano, F. Frare and G. Perugi, Pharmacopsychiatry, 1996, 29, 127–134 CrossRef CAS PubMed.
- (a) J. Knabe, H. P. Buech and S. Bender, Arch. Pharm., 1995, 328, 59–66 CrossRef CAS; (b) R. N. Brogden, R. C. Heel, T. M. Speight and G. S. Avery, Drugs, 1980, 20, 161–178 CrossRef CAS PubMed.
- A. Alija, M. Martin, S. Hartmut, K. Heinz, D. Sara, F. Mary, B. John and G. Andrew, PCT Int. Appl., 2003053967, 2003 Search PubMed.
- R. K. Verma, G. K. Verma, G. Shukla and M. S. Singh, RSC Adv., 2012, 2, 2413–2421 RSC.
- E. Heiner, P. Stefan and H. Silke, PCT Int. Appl., 2008145681, 2008 Search PubMed.
- R. G. Xing, Y. N. Li, Q. Liu, Q. Y. Meng, J. Li, X. X. Shen, Z. Liu, B. Zhou, X. Yao and Z. L. Liu, Eur. J. Org. Chem., 2006, 6627–6632 Search PubMed.
- M. A. Chari, D. Shobha, T. Sasaki, T. Lett., 2011, 52, 5575–5580.
- A. Benito, M. Manez, J. payfi, J. Soto, M. Tendero and E. Sinn, J. Organomet. Chem., 1995, 503, 259–263 CrossRef CAS.
- D. Saha, A. Saha and B. C. Ranu, Green Chem., 2009, 11, 733–737 RSC.
- B. M. Savall and J. Fontimayor, Tetrahedron Lett., 2008, 49, 6667–6669 CrossRef CAS PubMed.
- G. M. Coppola, Synth. Commun., 2008, 38, 3500–3507 CrossRef CAS.
- M. A. Charia, D. Shobha, E. R. Kenawyb, S. S. A. Deyab, B. V. S. Reddy and A. Vinu, Tetrahedron Lett., 2010, 51, 5195–5199 CrossRef PubMed.
- M. Shen and T. G. Driver, Org. Lett., 2008, 10, 3367–3370 CrossRef CAS PubMed.
- S. K. Xiang, W. Tan, D. X. Zhang, X. L. Tian, C. Feng, B. Q. Wang, K. Q. Zhao, P. Hu and H. Yang, Org. Biomol. Chem., 2013, 11, 7271 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of representative spectra. See DOI: 10.1039/c3ra47761d |
| This journal is © The Royal Society of Chemistry 2014 |