
Preparation of boron nitride and polystyrene/boron nitride composite particles by dehydrogenation in ionic liquids
Abstract
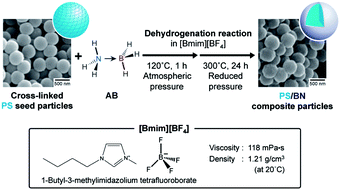
Boron nitride (BN) was prepared by the dehydrogenation of ammonia borane (AB) in an ionic liquid (1-butyl-3-methylimidazolium tetrafluoroborate, [Bmim][BF4]) at 300 °C, which is lower than the temperature of the general preparation method of BN and below the decomposition temperature of polystyrene (PS). The reaction was performed at 120 °C for 10 h under atmospheric pressure, and the product material was subsequently heated at 300 °C for 24 h under reduced pressure in [Bmim][BF4]. The reaction rate and final conversion increased when [Bmim][BF4] was used as the medium as compared to those observed in the bulk system (in the absence of the solvent system). Moreover, PS/BN composite particles were successfully prepared by dehydrogenation in [Bmim][BF4] in the presence of cross-linked PS seed particles. Transmission electron microscopy images of ultrathin cross-sections of the composite particles confirmed the core–shell morphology of the particles with a PS core and a BN shell.
 Please wait while we load your content...
Please wait while we load your content...

