Regioselective ortho-carboxylation of phenols catalyzed by benzoic acid decarboxylases: a biocatalytic equivalent to the Kolbe–Schmitt reaction†
Christiane Wuenschab,
Johannes Grossab,
Georg Steinkellnerac,
Andrzej Lyskowskiac,
Karl Gruberc,
Silvia M. Glueck*ab and
Kurt Faber*b
aAustrian Centre of Industrial Biotechnology (ACIB) GmbH, Petersgasse 14, A-8010 Graz, Austria
bDepartment of Chemistry, Organic & Bioorganic Chemistry, University of Graz, Heinrichstrasse 28, A-8010 Graz, Austria. E-mail: Si.Glueck@Uni-Graz.at; Kurt.Faber@Uni-Graz.at; Fax: +43-316-380-9840; Tel: +43-316-380-5332
cInstitute of Molecular Biosciences, University of Graz, Humboldtstrasse 50, A-8010 Graz, Austria
First published on 27th January 2014
Abstract
The enzyme catalyzed carboxylation of electron-rich phenol derivatives employing recombinant benzoic acid decarboxylases at the expense of bicarbonate as CO2 source is reported. In contrast to the classic Kolbe–Schmitt reaction, the biocatalytic equivalent proceeded in a highly regioselective fashion exclusively at the ortho-position of the phenolic directing group in up to 80% conversion. Several enzymes were identified, which displayed a remarkably broad substrate scope encompassing alkyl, alkoxy, halo and amino-functionalities. Based on the crystal structure and molecular docking simulations, a mechanistic proposal for 2,6-dihydroxybenzoic acid decarboxylase is presented.
Introduction
The sequestration of carbon from renewable resources to obtain value-added compounds has recently emerged as a prime goal in synthetic organic chemistry. A promising and especially environmentally friendly approach makes use of CO2 as a C1 building block thereby converting a problematic waste gas into useful well-defined products. However, since carbon in CO2 occurs in the highest possible oxidation state, it comes along with a low level of energy and makes its use as starting material for a chemical reaction very challenging, which explains why only a very limited number of carboxylation processes run on industrial scale.1,2 Various strategies were pursued in order to overcome the disfavored energetic balance of CO2 utilization such as the use of high energy starting materials, the transformation towards low-energy end products as well as additional input of energy or in situ (co)product removal.3,4Numerous chemical CO2 fixation concepts have been developed focusing on ‘green’ (homogenous and heterogeneous) catalytic processes, in which the functionalization of carbon dioxide proceed either through nucleophilic attack to form a carboxyl group or via the generation of a five-membered ring through oxidative cycloaddition,3 enabling sustainable methodologies for the production of carboxylic acids, urethane derivatives, esters, lactones, carbamates and (poly)carbonates or cyclic carbonates.2,3,5,6 Since the first transition-metal–CO2 complex [Ni(CO2)(PCy3)2] was reported,7 a considerable number of transition-metal catalysts were developed employing various metals as catalytic center (Ni, Pd, Rh, Cu, Au, Al, Zn, Fe).2,3,8,9 Among these, the carboxylation of aromatic C–H bonds represents an attractive synthetic tool to yield aromatic carboxylic acid derivatives. Several cross-coupling protocols have been reported, in which CO2 insertion occurs after initial deprotonation of an activated C–H bond by the metal complex. However, only comparatively acidic C–H bonds can be carboxylated at reasonable rates.6,8,9 Another approach represents the Rh-catalyzed direct carboxylation of arenes via chelation-assisted C–H activation applied to various 2-arylpyridines and 1-arylpyrazoles to afford the corresponding heteroaromatic carboxylic acids in up to 75% yield.6,8,10 In a similar fashion, the Pd-catalyzed direct carboxylation of aryl bromides11 and more recently, a Ni-catalyzed protocol for the carboxylation of aryl and vinyl chlorides has been demonstrated.12 An attractive alternative to (transition)-metal catalyzed CO2 fixation protocols makes use of organocatalysts which render environmentally compatible metal-free protocols.13,14
In contrast, nature provides a vast variety of enzymatic CO2-fixation reactions4 which play a vital role in all living organisms, most prominent RuBisCO (D-ribulose-1,5-bisphosphate carboxylase/oxygenase),15 which is one of the key enzymes in photosynthesis. Unfortunately, most of the biosynthetic CO2-fixing enzymes are highly substrate specific, which severely limits the chances to employ them for man-made ‘non-natural’ substrates. On the other hand, unspecific CO2-fixation occurs in biodegradation pathways, which serve as detoxification of electron-rich (hetero)aromatics in oxygen-depleted environments to enhance the water-solubility of toxic phenols.4 Whereas the oxidative degradation of aromatics predominantly involving mono-oxygenases has been well studied, only limited data are available on the anaerobic counterpart catalyzed by (de)carboxylases. In order to ensure fast turnovers and facile biochemical assays, the majority of enzymatic CO2-fixation reactions have been biochemically investigated in the energetically favored decarboxylation direction and the respective enzymes were commonly denoted as ‘decarboxylases’ rather than ‘carboxylases’.
The enzymatic carboxylation of electron-rich (hetero)aromatics can be divided into four major categories:
(1) Regioselective para-carboxylation4 of phenol exclusively occurs in the para-position to the directing phenolic group to furnish the corresponding para-hydroxybenzoic acids. The cofactor-independent carboxylation is catalyzed by 4-hydroxybenzoate-16–18 and 3,4-dihydroxybenzoate decarboxylases19,20 whereas phenyl phosphate carboxylases21,22 require an ATP-consuming activation of the phenol via phosphorylation prior to the carboxylation.
(2) Regiocomplementary ortho-carboxylation4 of phenol and its 1,2- and 1,3-dihydroxy analogues was described using 2,3-23,24 and 2,6-dihydroxybenzoate decarboxylases (also known as γ-resorcylate decarboxylases)25–27 and salicylic acid decarboxylases.28 The reaction proceeds in a highly regioselective fashion and affords the corresponding ortho-hydroxybenzoic acids exclusively.
(3) Electron-rich heteroaromatic compounds, such as pyrrole and indole, were enzymatically carboxylated in the α- and β-position by pyrrole-2-29,30 and indole-3-carboxylate decarboxylases,31 respectively.
(4) More recently, the enzymatic β-carboxylation of para-hydroxystyrenes was shown to proceed in a regiocomplementary fashion at the β-carbon atom of the styrene side chain yielding the corresponding (E)-cinnamic acids.32
Based on preliminary results,32 we report on the substrate scope of the ortho-carboxylation of benzoic acid decarboxylases from various microbial sources using a variety of structurally and electronically diverse non-natural substrates (Scheme 1). Previous reports focused on 'natural' substrates, such as pyrrole, indole, phenol and the corresponding 1,2- and 1,3-dihydroxy analogs. Only a single example delved into the ortho-carboxylation of m-aminophenol.33,34
 | ||
| Scheme 1 Regioselective ortho-carboxylation of phenol-type substrates employing benzoic acid decarboxylases to furnish the corresponding ortho-hydroxybenzoic acids. | ||
Results and discussion
Enzyme selection
In general, the genes encoding for (de)carboxylases which are described to catalyze the carboxylation of (hetero)aromatics (see above group 1–4) are distributed in all three microbial domains.35 Based on a search in protein databases (Brenda, NCBI, UniProt) with the focus on enzymes catalyzing the (de)carboxylation of phenolic substrates, a set of suitable enzyme candidates were chosen with emphasis on a broad phylogenetic diversity (for a phylogenetic tree see Fig. S1, ESI†). In order to obtain a reasonably spread selection of enzyme candidates, the sequence identity was kept within a range of 40–80% (see ESI, Table S1†), i.e. neither too high (to exclude isoenzymes possessing an identical substrate scope), nor too low (to avoid unrelated proteins). The following enzymes were chosen:(i) 2,3-Dihydroxybenzoic acid decarboxylase from Aspergillus oryzae (2,3-DHBD_Ao), (ii) salicylic acid decarboxylase from Trichosporon moniliiforme (SAD_Tm), (iii) 2,6-dihydroxybenzoic acid decarboxylases from Rhizobium sp. (2,6-DHBD_Rs) and three (iv) 4,5-dihydroxyphthalic acid decarboxylases from Bordetella pertussis (4,5-DHPD_Bp), Comamonas testosteroni (4,5-DHPD_Ct) and Verminephrobacter eiseniae (4,5-DHPD_Ve).
All genes were synthesized at Life Technologies (Germany) and subcloned in a pET vector (pET 21a). For overexpression a standard E. coli BL21(DE3) host was transformed with the particular plasmids using IPTG for induction. According to SDS-PAGE analysis, almost all enzymes were successfully overexpressed except 4,5-DHPD_Ve. For easier handling recombinant enzymes were applied as lyophilized whole cells. The absence of undesired side-activities by the host cells was verified in independent control experiments. In order to verify activities, initial tests were conducted in the energetically favored downhill decarboxylation direction using the ‘natural’ substrate. All enzyme candidates showed excellent activities indicated by full conversion for decarboxylation. For subsequent substrate screenings in the 'uphill' carboxylation direction, 2,3-DHBD_Ao, SAD_Tm and 2,6-DHBD_Rs were used. 4,5-DHPD's were not further investigated since they did not show any carboxylation activities towards their natural substrate.
Substrate spectrum
In order to examine the potential of the enzymatic ortho-carboxylation, a broad range of structurally and electronically diverse non-natural substrates were subjected to the enzymatic carboxylation using bicarbonate buffer as CO2-source (Table 1).27–29 For a complete overview, a few substrates previously published in a communication are included in Table 1.32 Overall, 2,3-DHBD_Ao and SAD_Tm displayed a remarkably broad substrate tolerance yielding the corresponding ortho-carboxylation products in up to 80% conversion. In contrast, 2,6-DHBD_Rs was somewhat restricted. The requirement for at least one phenolic hydroxyl group appeared as common feature of all three benzoic acid decarboxylases, which exclusively acted in the ortho-position of the 'directing' phenolic group. No trace of regioisomeric para-carboxylation products which often plagues the classic Kolbe–Schmitt reaction could be detected. With the exception of substrate 19a (Table 1, entries 19, 20), only a single regioisomeric carboxylation product was formed. In the absence of a free ortho-position (see ESI, Table S2,† entry 1), no reaction took place, the same is true for non-phenolic substrates (see ESI, Table S2† entries 2–8). A summary of non-substrates tested is given in the ESI, Table S2.†| Entry | Substrateb | Product | Enzymec | |||
|---|---|---|---|---|---|---|
| 1a–31a | 1b–31b | 2,3-DHBD_Ao | SAD_Tm | 2,6-DHBD_Rs | ||
| Subtype |
|
Conv.d [%] | Conv.d [%] | Conv.d [%] | ||
| a Reaction conditions: whole lyophilized cells (30 mg), substrate (10 mM), KHCO3 (3 M), phosphate buffer (pH 8.5, 100 mM), 30 °C, 120 rpm, 24 h.b Arrows indicate carboxylation site.c 2,3-DHBD_Ao = 2,3-dihydroxybenzoate decarboxylase from Aspergillus oryzae, 2,6-DHBD_Rs = 2,6-dihydroxybenzoate decarboxylase from Rhizobium sp. and SAD_Tm = salicylic acid decarboxylase from Trichosporon moniliiforme.d Conversion values refer to a single product, the only exception being substrate 19a, which yields two isomeric products (19b and 19c).e Data from ref. 32 are included for a complete overview. | ||||||
| 1 | 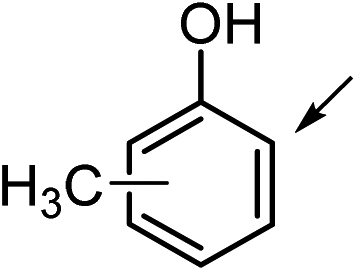 |
1a (R1 = CH3, R2 = R3 = H) | 1b | 16 | 30 | <1 |
| 2 | 2a (R2 = CH3, R1 = R3 = H) | 2b | 24 | 15 | 1 | |
| 3 | 3a (R3 = CH3, R1 = R2 = H) | 3b | 30 | 7 | 1 | |
| 4 |  |
4a (R1 = OMe, R2 = R3 = H) | 4b | 24 | 25 | <1 |
| 5 | 5a (R2 = OMe, R1 = R3 = H) | 5b | 50 | 48 | 12 | |
| 6 | 6a (R3 = OMe, R1 = R2 = H) | 6b | 1 | <1 | <1 | |
| 7 | 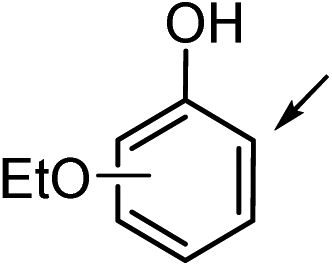 |
7a (R1 = OEt, R2 = R3 = H) | 7b | 25 | 26 | <1 |
| 8 | 8a (R2 = OEt, R1 = R3 = H) | 8b | 48 | 47 | 18 | |
| 9 | 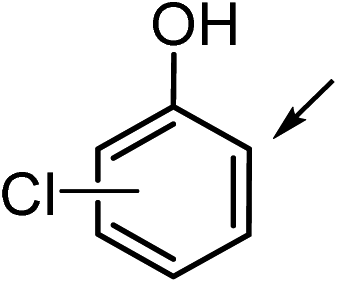 |
9a (R1 = Cl, R2 = R3 = H) | 9b | 19 | 17 | <1 |
| 10 | 10a (R2 = Cl, R1 = R3 = H) | 10b | 25 | 22 | 1 | |
| 11 | 11a (R3 = Cl, R1 = R2 = H) | 11b | 17 | 5 | 4 | |
| 12 |  |
12a (R1 = Br, R2 = R3 = H) | 12b | 18 | 21 | <1 |
| 13 | 13a (R2 = Br, R1 = R3 = H) | 13b | 13 | 14 | <1 | |
| 14 | 14a (R3 = Br, R1 = R2 = H) | 14b | 20 | 9 | 14 | |
| 15 |  |
15a (R1 = NH2, R2 = R3 = H) | 15b | 16 | 13 | <1 |
| 16 | 16ae (R2 = NH2, R1 = R3 = H) | 16b | 74 | 80 | 5 | |
| 17 | 17a (R3 = NH2, R1 = R2 = H) | 17b | <1 | <1 | <1 | |
| 18 |  |
18ae (R1 = OH, R2 = R3 = H) | 18b | 30 | 21 | 35 |
| 19 | 19ae (R2 = OH, R1 = R3 = H) | 19b | 22 | 30 | 31 | |
| 20 | 19c | 29 | 40 | <1 | ||
| 21 | 20a (R3 = OH, R1 = R2 = H) | 20b | <1 | <1 | <1 | |
| 22 |  |
21a (R1 = CH3) | 21b | 70 | 70 | 70 |
| 23 | 22ae (R1 = n-C5H11) | 22b | 39 | 2 | 56 | |
| 24 |  |
23ae (R1 = R2 = H) | 23b | 43 | 43 | <1 |
| 25 | 24a (R1 = H, R2 = CH2OH) | 24b | <1 | 26 | <1 | |
| 26 | 25a (R1 = OH, R2 = CH3) | 25b | 15 | 28 | 37 | |
| 27 |  |
26a (R1 = R2 = OH) | 26b | <1 | <1 | <1 |
| 28 | 27a (R1 = R2 = OMe) | 27b | 1 | 1 | <1 | |
| 29 |  |
28ae | 28b | 54 | 62 | 46 |
| 30 | 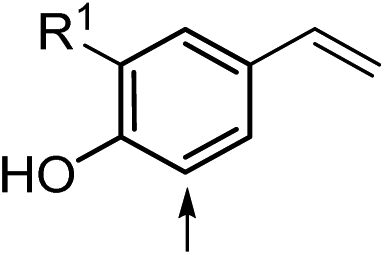 |
29ae (R1 = H) | 29b | 58 | 29 | <1 |
| 31 | 30ae (R1 = OMe) | 30b | 29 | 17 | <1 | |
| 32 |  |
31a | 31b | 14 | 5 | <1 |

Substituents in meta-position to the phenolic OH group (Table 1, entries 2, 5, 8, 10, 13, 16, 19, 20) were often (but not exclusively) obtained with higher conversion levels than the corresponding ortho- or para-analogs. para-Substituted substrates were often less favored (entries 3, 6, 11, 14, 17, 21). The electronic nature of the substituents did not have a significant impact on conversions, and alkyl, alkoxy, halo and amino groups (entries 1–17) were likewise tolerated, depending on the enzyme. The only exception were substrates bearing strong e−-withdrawing substituents (e.g. NO2 or carbonyl group), which were not carboxylated at all (see ESI, Table S2,† entries 9–11). Steric restrictions were not evident, since the differences between methoxy- and ethoxy-analogs were small (entries 4–6 vs. 7, 8) and even the n-pentyl substituted compound 22a was nicely converted (entry 23). In contrast, an extension of an ortho-alkyl moiety was not accepted (Table S2,† entries 12, 13). With a higher degree of substitution, the picture changed: whereas some doubly substituted phenols (entries 22, 23, 26) were carboxylated at acceptable rates, 3,5-dihydroxy- (26a) and 3,5-dimethoxyphenol (27a) (entries 27, 28), as well as m-substituted catechols were not converted (Table S2,† entries 14, 15). Surprisingly, also α-naphthol (28a) was selectively carboxylated in the β-position in up to 62% conversion (entry 29), while β-naphthol (47a), 7,8-dihydronaphthalen-2-ol (48a) and 5,6,7,8-tetrahydronapthalene-1-ol (49a) were non-substrates (Table S2,† entries 16–18). An interesting electronic effect emerged with substrates 30a and 31a (entries 31, 32): while conjugated 2-methoxy-4-vinylphenol was reasonably accepted (cmax 29%), the non-conjugated p-allyl analog was a poor substrate (cmax 14%).
Upscaling
In order to prove the applicability of the method on preparative scale, the ortho-carboxylation was scaled up using orcinol (21a) as model substrate. Almost identical conversions (67 and 68%, respectively) were obtained when the amount of substrate was raised from 10 mM (4.3 mg) to 50 mM (21.3 mg), whereas a further increase [100 mM (42.7 mg) and 235 mM (100 mg)] led to a drop in conversion (23% and 4%, respectively). A substrate concentration study using resorcinol (19a) was demonstrated recently.36Overall, the enzymatic ortho-carboxylation of phenols represents a promising biocatalytic equivalent to the classical (chemical) Kolbe–Schmitt reaction37 to furnish the corresponding 2-hydroxycarboxylic acids. For example: the tuberculostatic agent p-aminosalicylic acid 16b (from m-aminophenol 16a) was obtained in up to 80% conversion/yield applying the enzymatic32,33 or chemical37 strategy. However, the chemical route requires high pressure and temperature whereas the enzyme-catalyzed counterpart proceeds at ambient and eco-friendly conditions (room temperature, aqueous buffer solution, atmospheric pressure, enzyme as catalyst). For resorcinol (19a) as substrate, the chemical route is plagued by low regioselectivity (furnishing a mixture of ortho- and para-products), whereas the ‘Bio-Kolbe–Schmitt’ variant employing 2,6-DHBD_Rs is distinguished by excellent regioselectivity yielding a single product isomer.
Proposal for catalytic mechanism
The presence of a catalytically active Zn2+ in the active site of 2,6-dihydroxybenzoate decarboxylase from Rhizobium sp.38 invokes a striking resemblance to the metal-dependent Kolbe–Schmitt reaction, where the regioselectivity of carboxylation can be switched from the ortho- to the para-position by replacing the small Na+ by the larger K+. The crystal structure of this enzyme has been solved by Goto et al.38 (Strain MTP-1005, PDB code 2DVU) and the authors also proposed a catalytic mechanism for the decarboxylation using 2,6-dihydroxybenzoate as model substrate. The proposed oxyanion intermediate of the carboxylation product 2,6-dihydroxybenzoic acid (19b) was docked into the active site and the obtained model was further refined by energy minimization of the selected binding modes (Fig. 1). From this arrangement, it can be concluded that the carboxylation is initiated by proton abstraction of the phenolic hydroxyl group through Asp287 activated in a triad by His218 and Glu221 (Scheme 2). This enhances the nucleophilicity of the ortho-carbon and enables a nucleophilic attack onto bicarbonate, which is coordinated to Zn2+ which itself is tightly chelated by Asp287, Glu8, His10 and His164. The oxy-anion intermediate thus formed is re-aromatized by the aid of a network of three catalytically important and structurally conserved water molecules (W1, W2, W3), triggered by Asn128 via a protonation-deprotonation sequence. Finally, the carboxylated product is released from the active site and is replaced by another substrate for a subsequent catalytic cycle. | ||
| Fig. 1 Active site representation of 2,6-DHBD_Rs. (A): Crystal structure of 2,6-DHBD_Rs (PDB-Code: 2DVU) with 2,6-dihydroxybenzoic acid (19b) (blue) in the active site. (B): Energy minimized docking of the proposed oxyanion intermediate. Water molecules are represented as red spheres. The active site cavity is shown as transparent surface and colored by hydrophobicity ranging from hydrophobic (red) via green to hydrophilic (blue). Dashed black lines show the coordination of Zn2+ and the hydrogen bonding network. Figure was prepared by using PyMOL.39 | ||
 | ||
| Scheme 2 Proposed catalytic mechanism for the ortho-carboxylation of substrate 19a based on the crystal structure of 2,6-dihydroxybenzoate decarboxylase from Rhizobium sp.38 (PDB-Code: 2DVU) supported by docking experiments. | ||
Conclusion
A broad range of phenol derivatives bearing alkyl, alkoxy, halo and amino-functionalities were successfully carboxylated using cloned and overexpressed benzoic acid decarboxylases. In contrast to the classic Kolbe–Schmitt reaction, the biocatalytic equivalent proceeded with exquisite regioselectivity at the ortho-position of the directing phenolic group under ambient reaction conditions using bicarbonate as CO2 source in up to 80% conversion.Experimental section
General
All NMR experiments were performed either on a Bruker Avance III 300 MHz spectrometer using a 5 mm BBO probe or on a Bruker Avance III 500 MHz spectrometer using a 5 mm TXI probe with z-axis gradients at 300 K, shifts are reported in ppm. Autoclave experiments were carried out in a mini reactor system (series 4560) from Parr Instrument Company.Enzymes
2,3-Dihydroxybenzoic acid decarboxylase from Aspergillus oryzae (2,3-DHBD_Ao), salicylic acid decarboxylase from Trichosporon moniliiforme (SAD_Tm) and 2,6-dihydroxybenzoic acid decarboxylase from Rhizobium species (2,6-DHBD_Rs) were synthesized and ligated into a pET 21a (+) vector at Life Technologies (Germany). Their DNA sequences were identified by sequence comparison in the NCBI Genebank (GI: 94730373 for 2,3-DHBD_Ao, GI: 225887918 for SAD_Tm and GI: 116667102 for 2,6-DHBD_Rs). Cloning and overexpression was performed as previously described.32General procedure for the carboxylation of substrates 1a–31a
For carboxylation reactions lyophilized whole cells (30 mg E. coli host cells containing the corresponding overexpressed enzyme) were resuspended in phosphate buffer (1 mL, pH 5.5, 100 mM) and were rehydrated for 30 min. The cell suspension was transferred to glass vials followed by addition of substrate (10 mM final concentration) and KHCO3 (3 M, 300 mg, final pH 8.5). In order to avoid loss of CO2, the vials were immediately tightly closed using a septum and the mixture was shaken for 24 h at 30 °C and 120 rpm. Thereafter the mixture was centrifuged (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 15 min) and an aliquot of 100 μL was diluted with an H2O–acetonitrile mixture (1 mL, v/v 50%) supplemented with trifluoroacetic acid (3% v/v, 30 μL) to quench the reaction. The samples were incubated at room temperature for 5 min and centrifuged (13000 rpm, 15 min). The conversion was analyzed by reverse-phase HPLC. Products were identified by co-injection with authentic reference material and the absence of undesired side-activities by the host cells was verified in independent control experiments using lyophilized empty E. coli host cells.
000 rpm, 15 min) and an aliquot of 100 μL was diluted with an H2O–acetonitrile mixture (1 mL, v/v 50%) supplemented with trifluoroacetic acid (3% v/v, 30 μL) to quench the reaction. The samples were incubated at room temperature for 5 min and centrifuged (13000 rpm, 15 min). The conversion was analyzed by reverse-phase HPLC. Products were identified by co-injection with authentic reference material and the absence of undesired side-activities by the host cells was verified in independent control experiments using lyophilized empty E. coli host cells.
For upscaling, the standard screening procedure was employed with lyophilized whole cells (100 mg) at 50 mM substrate concentration in a 3 mL final volume of phosphate buffer.
Synthesis of reference material
For the synthesis of reference material for 22b, 29b and 30b see ref. 32.Analytical procedures
Method A was run over 35 min with H2O–trifluoroacetic acid (0.1%) as the mobile phase (flow rate 0.5 mL min−1) and an acetonitrile–trifluoroacetic acid (0.1%) gradient (0–2 min 0%, 2–15 min 0–32%, 15–20 min 32%, 20–35 min 32–100%). Method B was run over 35 min with H2O–trifluoroacetic acid (0.1%) as the mobile phase (flow rate 0.5 mL min−1) and an acetonitrile–trifluoroacetic acid (0.1%) gradient (0–2 min 0%, 2–30 min 0–100%, 30–35 min 100%). Method C was run over 10 min with phosphate buffer (0.12 M, pH 2.4) as the mobile phase (flow rate 0.5 mL min−1). Method D was run over 10 min with H2O–acetonitrile (50/50) supplemented with trifluoroacetic acid (0.1%) as the mobile phase (flow rate 0.5 mL min−1). Method E was run over 12 min with H2O–trifluoroacetic acid (0.1%) as the mobile phase (flow rate 1 mL min−1) and an acetonitrile–trifluoroacetic acid (0.1%) gradient (0–2 min 15%, 2–10 min 15–100%, 10–12 min 100%). Method F was run over 17 min with H2O–trifluoroacetic acid (0.1%) as the mobile phase (flow rate 1 mL min−1) and an acetonitrile–trifluoroacetic acid (0.1%) gradient (0–2 min 0%, 2–15 min 0–100%, 15–17 min 100%). Method G was run over 10 min with H2O–acetonitrile (50/50) supplemented with trifluoroacetic acid (0.1%) as the mobile phase (flow rate 1 mL min−1). Method H was run over 55 min with phosphate buffer (0.12 M, pH 2.4) as the mobile phase (flow rate 0.5 mL min−1). For retention times of substrates and products see Table S3 in the ESI.†
Acknowledgements
This work was supported by the Austrian BMWFJ, BMVIT, SFG, Standortagentur Tirol and ZIT through the Austrian FFG-COMET-Funding Program. Klaus Zangger and his coworkers (University of Graz) are thanked for their valuable support in NMR-spectroscopy.References
- E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- S. M. Glueck, S. Gümüs, W. M. F. Fabian and K. Faber, Chem. Soc. Rev., 2010, 39, 313–328 RSC.
- D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780 CrossRef CAS PubMed.
- R. Martin and A. W. Kleij, ChemSusChem, 2011, 4, 1259–1264 CrossRef CAS PubMed.
- M. Aresta and C. F. Nobile, J. Chem. Soc., Chem. Commun., 1975, 636–637 RSC.
- Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC.
- K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC.
- H. Mizuno, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 1251–1253 CrossRef CAS PubMed.
- A. Correa and R. Martin, J. Am. Chem. Soc., 2009, 131, 15974–15975 CrossRef CAS PubMed.
- T. Fujihara, K. Nogi, T. Xu, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2012, 134, 9106–9109 CrossRef CAS PubMed.
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032–2038 CrossRef CAS PubMed.
- A. Ueno, Y. Kayaki and T. Ikariya, Green Chem., 2013, 15, 425–430 RSC.
- G. Schneider, Y. Lindqvist and C.-I. Brändern, Annu. Rev. Biophys. Biomol. Struct., 1992, 21, 119–143 CrossRef CAS PubMed.
- J. Huang, Z. He and J. Wiegel, J. Bacteriol., 1999, 181, 5119–5122 CAS.
- J. Liu, X. Zhang, S. Zhou, P. Tao and J. Liu, Curr. Microbiol., 2007, 54, 102–107 CrossRef CAS PubMed.
- T. Matsui, T. Yoshida, T. Hayashi and T. Nagasawa, Arch. Microbiol., 2006, 186, 21–29 CrossRef CAS PubMed.
- Z. He and J. Wiegel, J. Bacteriol., 1996, 178, 3539–3543 CAS.
- T. Yoshida, Y. Inami, T. Matsui and T. Nagasawa, Biotechnol. Lett., 2010, 32, 701–705 CrossRef CAS PubMed.
- M. Aresta, E. Quaranta, R. Liberio, C. Dileo and I. Tommasi, Tetrahedron, 1998, 54, 8841–8846 CrossRef CAS.
- M. Boll and G. Fuchs, Biol. Chem., 2005, 386, 989–997 CAS.
- A. V. Kamath, D. Dasgupta and C. S. Vaidyanathan, Biochem. Biophys. Res. Commun., 1987, 145, 586–595 CrossRef CAS.
- R. Santha, N. Appaji Rao and C. S. Vaidyanathan, Biochim. Biophys. Acta, 1996, 1293, 191–200 CrossRef.
- M. Yoshida, N. Fukuhara and T. Oikawa, J. Bacteriol., 2004, 186, 6855–6863 CrossRef CAS PubMed.
- Y. Iwasaki, K. Kino, H. Nishide and K. Kirimura, Biotechnol. Lett., 2007, 29, 819–822 CrossRef CAS PubMed.
- T. Matsui, T. Yoshida, T. Yoshimura and T. Nagasawa, Appl. Microbiol. Biotechnol., 2006, 73, 95–102 CrossRef CAS PubMed.
- K. Kirimura, H. Gunji, R. Wakayama, T. Hattori and Y. Ishii, Biochem. Biophys. Res. Commun., 2010, 394, 279–284 CrossRef CAS PubMed.
- M. Wieser, T. Yoshida and T. Nagasawa, J. Mol. Catal. B: Enzym., 2001, 11, 179–184 CrossRef CAS.
- T. Yoshida and T. Nagasawa, J. Biosci. Bioeng., 2000, 89, 111–118 CrossRef CAS.
- T. Yoshida, K. Fujita and T. Nagasawa, Biosci., Biotechnol., Biochem., 2002, 66, 2388–2394 CrossRef CAS.
- C. Wuensch, S. M. Glueck, J. Gross, D. Koszelewski, M. Schober and K. Faber, Org. Lett., 2012, 14, 1974–1977 CrossRef CAS PubMed.
- K. Kirimura, S. Yanaso, S. Kosaka, K. Koyama, T. Hattori and Y. Ishii, Chem. Lett., 2011, 40, 206–208 CrossRef CAS.
- S. Ienaga, S. Kosaka, Y. Honda, Y. Ishii and K. Kirimura, Bull. Chem. Soc. Jpn., 2013, 86, 628–634 CrossRef CAS.
- B. Lupa, D. Lyon, M. D. Gibbs, R. A. Reeves and J. Wiegel, Genomics, 2005, 86, 342–351 CrossRef CAS PubMed.
- C. Wuensch, N. Schmidt, J. Gross, B. Grischek, S. M. Glueck and K. Faber, J. Biotechnol., 2013, 168, 264–270 CrossRef CAS PubMed.
- A. S. Lindsey and H. Jeskey, Chem. Rev., 1957, 57, 583–620 CrossRef CAS.
- M. Goto, H. Hayashi, I. Miyahara, K. Hirotsu, M. Yoshida and T. Oikawa, J. Biol. Chem., 2006, 281, 34365–34373 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC Search PubMed.
- D. Cameron, M. Jeskey and O. Baine, J. Org. Chem., 1950, 15, 233–236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47719c |
| This journal is © The Royal Society of Chemistry 2014 |