DOI:
10.1039/C3RA47718E
(Paper)
RSC Adv., 2014,
4, 9608-9616
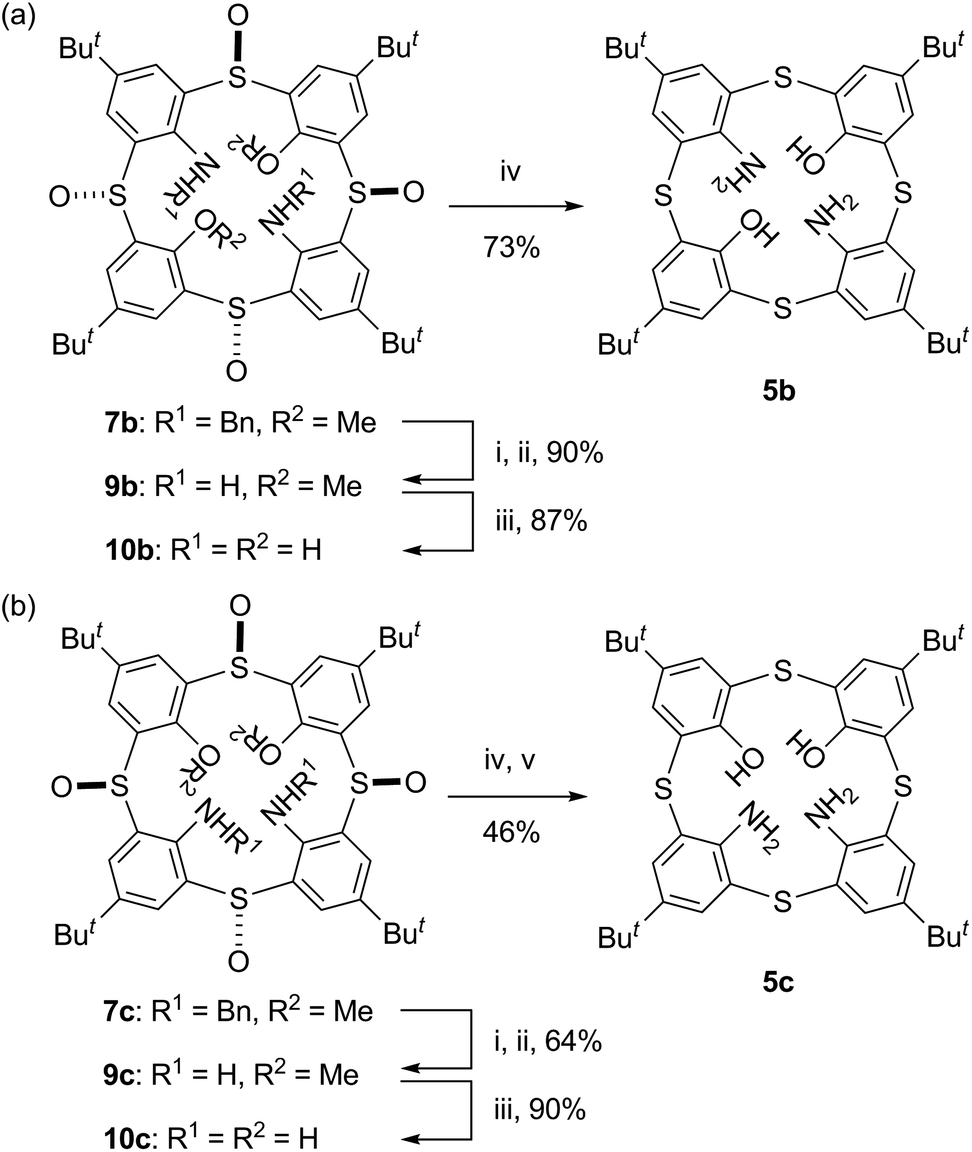
Regioselective synthesis of 1,2- and 1,3-diaminothiacalix[4]arenes via nucleophilic aromatic substitution and their X-ray structures†
Received
17th December 2013
, Accepted 27th January 2014
First published on 28th January 2014
Abstract
Thiacalix[4]arenes bearing two amino groups instead of two hydroxy groups of p-tert-butylthiacalix[4]arene (2) at the distal 1,3- (5b) and proximal 1,2-positions (5c) are regioselectively synthesized via the nucleophilic aromatic substitution (SNAr) reaction of lithium benzylamide with tetra-O-methylsulfinylcalix[4]arenes 4(rctt) and 4(rcct), respectively. The SNAr reaction selectively proceeds at the benzene rings between two neighboring sulfinyl groups of a trans conformation. This is rationalized by the cooperative coordination of a methoxy group with a neighboring sulfinyl oxygen atom toward the metal ion to form a six-membered chelate, through which the amide ion attacks the aromatic nucleus. The N,O-hybrid thiacalixarenes 5b and 5c adopt cone conformations in their acetonitrile inclusion crystals. The distances between adjacent N and/or O atoms of their narrow-rim substituents indicate that the four substituents form a circular hydrogen-bond network but it is weaker than that of original thiacalixarene 2. Compound 5c also adopts a cone conformation in the guest-free crystal. This, combined with the fact that tetraaminated p-tert-butylthiacalix[4]arene 5a adopts a 1,3-alternate conformation in its guest-free crystal, indicates that the intramolecular O⋯N hydrogen bonds, not to mention the O⋯O hydrogen bond, in compound 5c play a crucial role in maintaining a cone conformation in the crystal.
Introduction
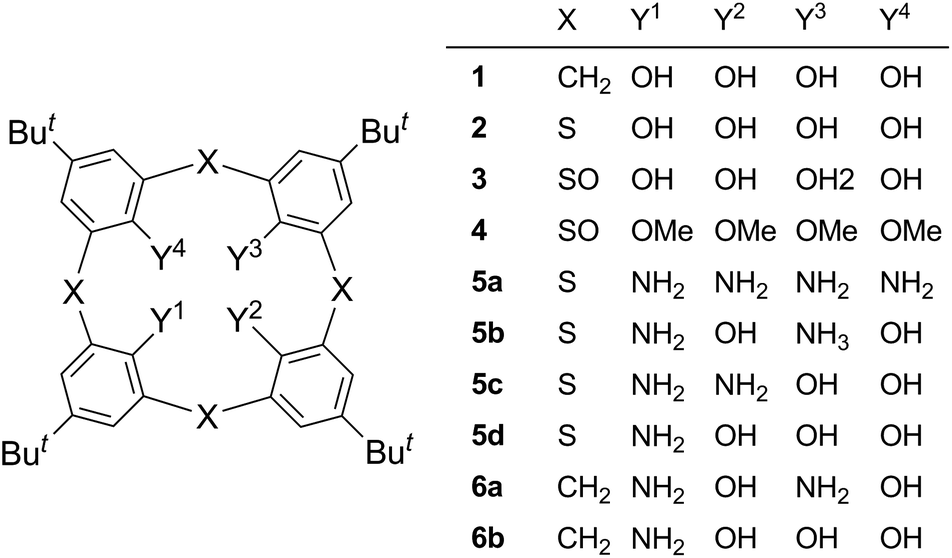
Calix[4]arenes (e.g. 1)1 and thiacalix[4]arenes (e.g. 2)2 are cyclic phenol tetramers in which adjacent phenol units are bridged at the ortho–ortho positions by methylene and sulfur moieties, respectively. They are extensively utilized as molecular scaffolds in host–guest supramolecular chemistry. In order to elaborate sophisticated hosts, a variety of functional groups have been introduced on the hydroxy groups through etherification or esterification and/or at the para positions to the hydroxy groups via electrophilic aromatic substitution reactions. On the other hand, since it is difficult to directly construct calix macrocycles from aromatic compounds other than phenol (e.g. aniline), synthesis of derivatives such as tetraaminothiacalixarene 5a has to rely on the displacement of the hydroxy groups of phenol-based calixarenes via the cleavage of the aryl–oxygen bonds. However, successful examples for such transformations are quite limited in the literature.3–7 It is known that an alkoxy group on an aromatic nucleus is readily displaced with a nucleophile via the conjugate activation by an electron-withdrawing group at the ortho position to the alkoxy group.8,9 The most notable reaction of this type is Meyers' oxazolinyl group-mediated one.10 In our previous paper, we succeeded in the preparation of tetraaminothiacalixarene 5a by applying this chelation-assisted nucleophilic aromatic substitution (SNAr) protocol to the benzylamination of a stereoisomer of tetra-O-methylsulfinylcalixarene 4(rtct).4d The displacement of the hydroxy groups of thiacalixarene 2 with amino groups (5a) noticeably changed affinity toward metal ions; in a solvent extraction experiment, tetraamine 5a selectively extracted Au(III) and Pd(II) ions,11 whereas tetraol 2 extracted a variety of metal ions ranging from soft to intermediate, depending on the soft/hard nature of the binding moieties.12,13 This finding prompted us to explore synthetic methods of hybrid-type thiacalixarenes 5b–d, which have both aniline and phenol units in the same framework. In our continuing efforts along this line, we have recently succeeded in preparing monoamine 5d and 1,3-diamine 5b via the Ullmann-type condensation of the 1,3-bistriflate ester of compound 2 with benzylamine and tosylamide, respectively.4e However, the synthesis of 1,2-diamine 5c has yet to be achieved. In this paper, we report the regioselective synthesis of 1,3-diamine 5b and 1,2-diamine 5c via the chelation-assisted SNAr reaction.14 The regioselectivity is discussed based on the chelation-assisted mechanism. Also reported is the effect of the amino groups on the circular hydrogen-bond network among the narrow-rim substituents of compounds 5b and 5c as studied by X-ray crystallographic analysis.
Results and discussion
Benzylamination of tetra-O-methylsulfinylcalixarenes 4
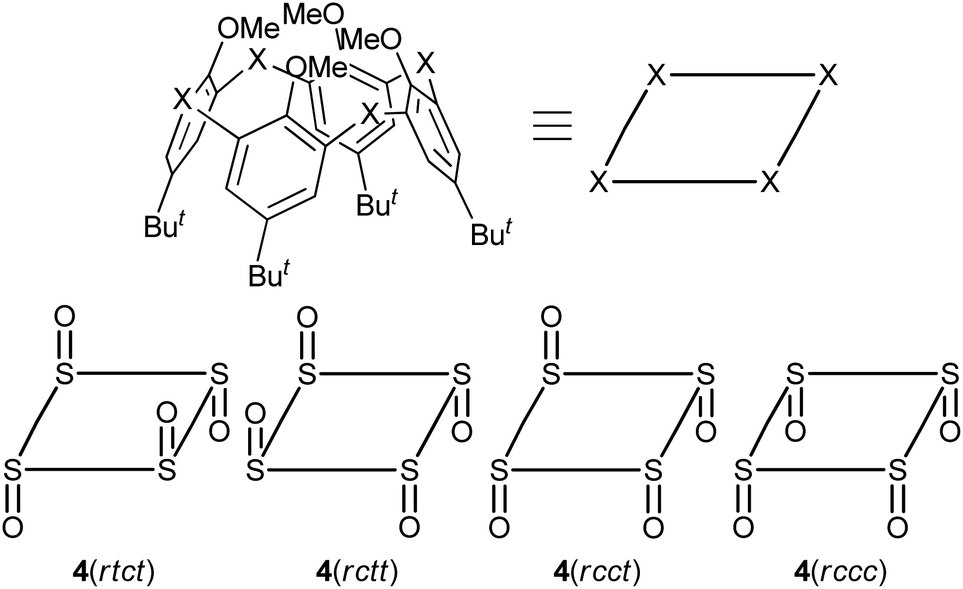
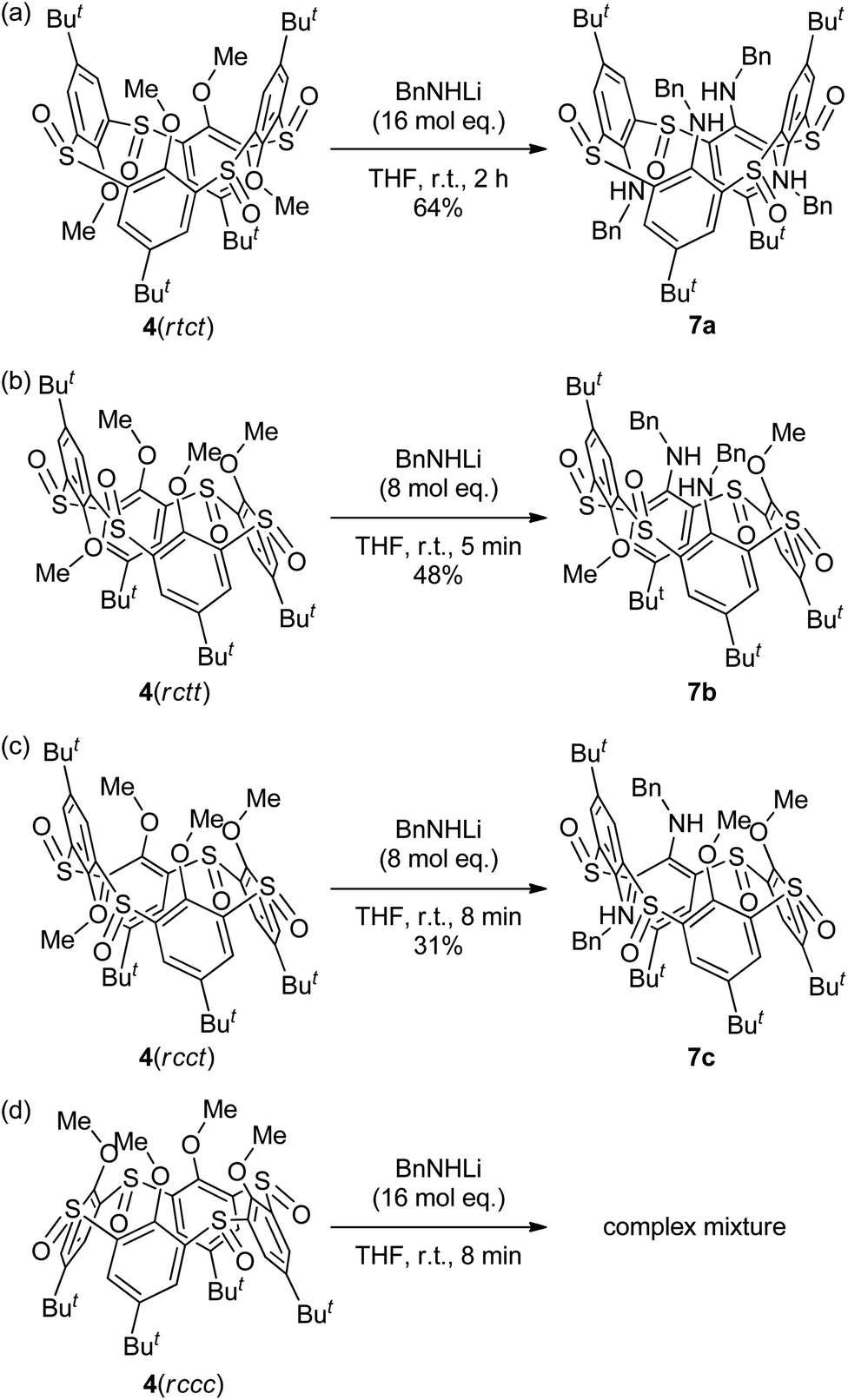
Tetra-O-methylsulfinylcalixarene 4 has four stereoisomers (rtct, rctt, rcct and rccc) originating from the arrangement of the four sulfinyl functions (Fig. 1). Previously, we succeeded in the preparation of all the stereoisomers15 and found that they exhibit different conformational behaviours in solution (vide infra).16 We imagined that such conformational differences might affect the regioselectivity of benzylamination. In a previously paper,4d we reported that isomer 4(rtct), on treatment with 16 mol equiv. of lithium benzylamide in THF for 2 h, gave tetra(benzylamino)sulfinylcalixarene 7a with a 1,3-alternate conformation (Scheme 1a). Therefore, isomer 4(rctt) was first submitted to the benzylamination under the same conditions but for a shorter time (30 min). However, the reaction gave a complex mixture, from which no identifiable compound could be isolated. The SNAr reaction of compound 4 with a nucleophile often competes with the nucleophilic reaction of the reagent to the sulfinyl functions. Actually, the reaction of compound 4(rtct) with butyllithium gave a ring-opened product, 4-tert-butyl-2-(butylsulfinyl)-1-methoxybenzene (8) (Scheme S1†),17 whereas the reaction with lithium benzylamide gave methoxy-substitution product 7a (vide supra). Therefore, one possible reason for the formation of no SNAr product in the benzylamination of isomer 4(rctt) was that the reagent preferentially reacted with the sulfinyl functions. To our delight, when the reaction was conducted using a reduced amount (8 mol equiv.) of lithium benzylamide for a shorter time (5 min), di(benzylamine) 7b was obtained in a 48% yield (Scheme 1b). A close inspection of other products by TLC combined with 1H NMR spectroscopy revealed that tri- and tetraaminated compounds, as well as regioisomers of compound 7b, were not formed at all. Under similar conditions, isomer 4(rcct) gave di(benzylamine) 7c in a 31% yield (Scheme 1c). Modification of the reaction time and/or the molar equivalence of lithium benzylamide did not improve the yields of the di(benzylamine)s. On the other hand, the reaction of isomer 4(rccc) gave a complex mixture, regardless of the reaction time, temperature and molar equivalence of the nucleophile (Scheme 1d).
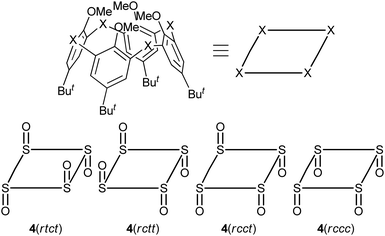 |
| | Fig. 1 Schematic views of stereoisomeric compounds 4. The dispositions of the sulfinyl oxygen atoms are denoted by the term cis (c) or trans (t) that is relative to the reference sulfinyl oxygen atom (r) with respect to the mean plane defined by the four sulfur atoms as suggested by Böhmer.1f The notation proceeds around the system in a direction from the reference oxygen, which is chosen in order to maximize the number of cis and prioritize cis over trans. | |
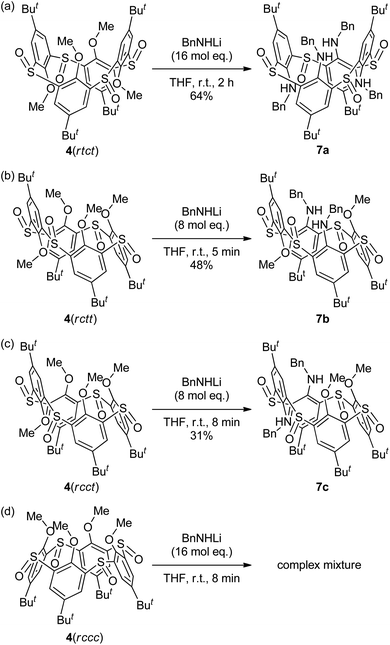 |
| | Scheme 1 Chelation-assisted SNAr reaction of compounds 4. (a) Data quoted from ref. 4d. | |
Determination of the regio- and stereochemistry of di(benzylamine)s 7b and 7c
The 1H NMR spectrum of di(benzylamine) 7b exhibited three singlets with an integrated intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 for the tert-butyl protons, suggesting that the compound has a symmetric structure with a σ-plane. The –CH2NH– protons appeared as an ABX pattern (2H each), whereas the methoxy protons appeared as two singlets (3H each). This indicates that the two benzylamino groups are magnetically equivalent and the methoxy groups are not. These observations unambiguously assigned this compound to be syn-1,3-dibenzylamine 7b, in which two benzylamino groups are attached to the benzene rings between two neighboring sulfinyl groups of a trans conformation; we denote hereafter such a benzene unit between two neighboring sulfinyl groups of a trans conformation by a trans-benzene unit and a benzene unit between two neighboring sulfinyl groups of a cis conformation by a cis-benzene unit. Although the orientation of the methoxy groups cannot be deduced from the 1H NMR spectrum, they should be oriented anti to the adjacent sulfinyl oxygen atoms to minimize electrostatic repulsion (vide infra).
:1:2 for the tert-butyl protons, suggesting that the compound has a symmetric structure with a σ-plane. The –CH2NH– protons appeared as an ABX pattern (2H each), whereas the methoxy protons appeared as two singlets (3H each). This indicates that the two benzylamino groups are magnetically equivalent and the methoxy groups are not. These observations unambiguously assigned this compound to be syn-1,3-dibenzylamine 7b, in which two benzylamino groups are attached to the benzene rings between two neighboring sulfinyl groups of a trans conformation; we denote hereafter such a benzene unit between two neighboring sulfinyl groups of a trans conformation by a trans-benzene unit and a benzene unit between two neighboring sulfinyl groups of a cis conformation by a cis-benzene unit. Although the orientation of the methoxy groups cannot be deduced from the 1H NMR spectrum, they should be oriented anti to the adjacent sulfinyl oxygen atoms to minimize electrostatic repulsion (vide infra).
Unfortunately, the regio- and stereochemistry of di(benzylamine) 7c could not be determined from its 1H NMR spectrum, which exhibited four singlets (9H each) for the tert-butyl protons, two singlets (3H each) for the methoxy protons, in addition to two sets of signals for the benzylamino groups; this fully unsymmetrical resonance pattern only excluded Cs-symmetric syn-1,2-dibenzylamines with cone and 1,2-alternate conformations, in which both the benzylamino moieties are attached to the trans-benzene units or, alternatively, the cis-benzene units. We then tried X-ray crystallographic analysis. Prerequisite single crystals were obtained by slow diffusion of methanol vapor to a chloroform solution of compound 7c. The X-ray analysis revealed that the two benzylamino groups are attached to the trans-benzene units and disposed anti to each other (Fig. S1†). The methoxy groups are in the direction opposite to the two neighboring sulfinyl oxygen atoms.
Mechanistic consideration of the regioselectivity of benzylamination
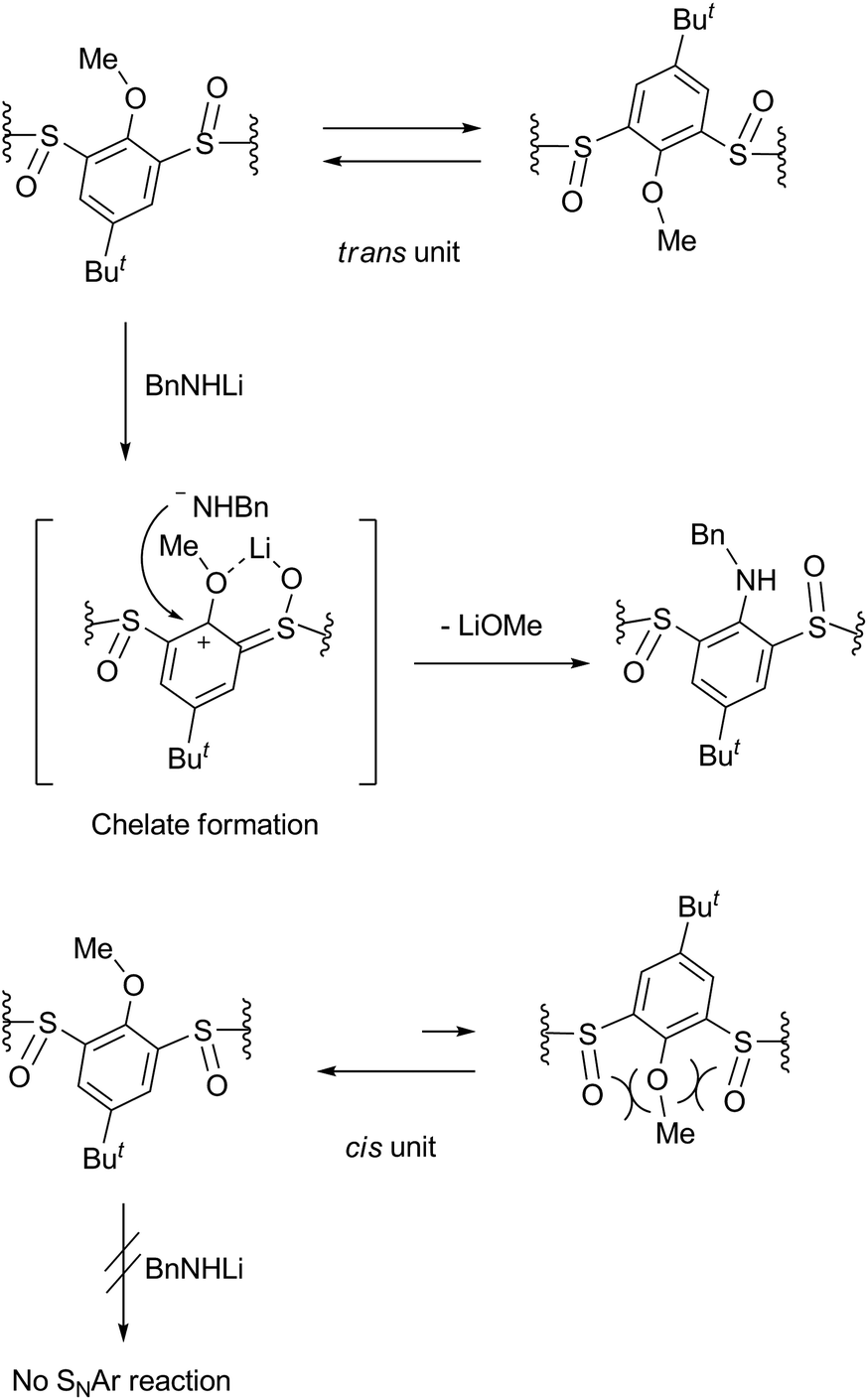
The benzylamination of four stereoisomers 4 shares a common characteristic with regard to the regioselectivity. Thus, all the methoxy groups attached to the trans-benzene units were displaced by the lithium amide, whereas any methoxy groups attached to the cis-benzene units were not. Previously, we examined the conformational behaviours of compounds 4 by VT NMR spectroscopy.16 It revealed that the rtct-isomer adopts a 1,3-alternate conformation and the rctt- and rcct-isomers adopt partial-cone conformations as shown in Scheme 1. All the trans-benzene units of each compound make a flip-flop motion simultaneously, while the cis-benzene units are fixed directing the methoxy groups opposite to the neighboring sulfinyl oxygen atoms to minimize dipole repulsion between the methoxy and sulfinyl groups. As a result, each compound exhibits a self-exchange between two equivalent species. On the other hand, the rccc-isomer is in an equilibrium state between cone (Scheme 1) and partial-cone conformers through the flip-flop motion of a benzene ring; the molar ratio is 85:15 (cone:partial cone) in CDCl3 at 253 K. The regioselectivity of the benzylamination can be rationalized by the conformational behaviours of compounds 4 (Scheme 2). The methoxy groups attached to the trans-benzene units are inevitably in the same direction to one of the neighboring sulfinyl oxygen atoms regardless of the self-exchange. Therefore, they can readily form six-membered chelates with lithium benzylamide in cooperation with the sulfinyl oxygen atoms in the same direction. The SNAr reaction proceeds through the chelates. On the other hand, the cis-benzene units of the rctt-, rcct- and rccc-isomers necessitate a conformational change to a higher-energy state to form such chelate complexes because the methoxy groups are in the opposite direction to both the neighboring sulfinyl oxygen atoms. As a result, the benzylamination selectively proceeds at the trans-benzene units. Based on this mechanism, the rccc-isomer may undergo benzylamination through the partial-cone conformer. However, the reaction gave no aminated calixarenes. As mentioned earlier, the SNAr reaction competes with the decomposition of the substrate through the nucleophilic reaction of the amide reagent to the sulfinyl functions. The abundance of the partial-cone conformer (15%) seems to be insufficient to overcome the decomposition.
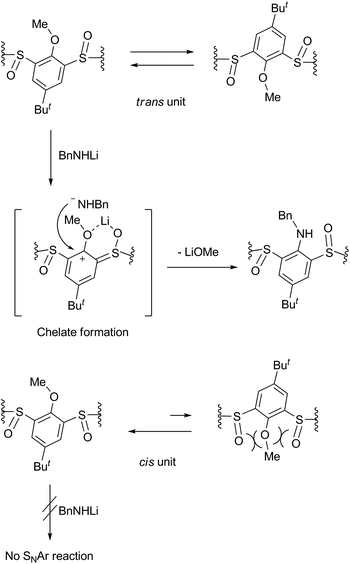 |
| | Scheme 2 Reaction mechanism that explains the regioselectivity of the benzylamination. | |
Deprotection and reduction of di(benzylamine)s 7b and 7c to N,O-hybrid thiacalixarenes 5b and 5c
In order to access to N,O-hybrid thiacalixarenes 5b and 5c, di(benzylamine)s 7b and 7c were subjected to a deprotection/reduction sequence (Scheme 3), which we previously employed for the conversion of tetra(benzylamine) 7a into tetraaminothiacalixarene 5a.4d Thus, the benzylamino moieties of compounds 7 were brominated with N-bromosuccinimide (NBS) and dibenzoyl peroxide (BPO). The reaction was followed by spontaneous dehydrobromination to give unstable imines, which were hydrolyzed without purification. The resulting free diamines 9 were demethylated with octane-1-thiolate and then reduced with LiAlH4 and TiCl4 to furnish N,O-hybrid thiacalixarenes 5b and 5c. To the best of our knowledge, the latter compound is the first X,O-hybrid calix[4]arene bearing two X groups at the proximal positions except for 1,2-hydrogenated calix[4]arene.18
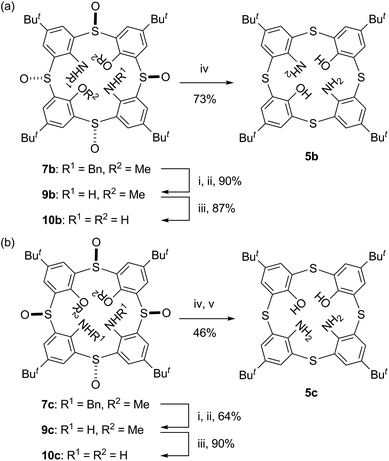 |
| | Scheme 3 Reagents and conditions: (i) NBS, BPO, benzene, reflux; (ii) 6 M HCl, CHCl3, reflux; (iii) CH3(CH2)7SNa, THF, reflux; (iv) LiAlH4, TiCl4, THF, r.t.; (v) Bu4NF, THF, r.t. | |
Conformations of N,O-hybrid thiacalixarenes 5b and 5c in the crystals
Thiacalixarene 2 adopts a cone conformation in a solution by virtue of a circular network of intramolecular hydrogen bonds between the hydroxy groups and accommodates various small organic molecules into its cavity to form inclusion crystals.19 In a guest-free crystal of compound 2, a pair of calixarenes with a cone conformation include each other's tert-butyl groups into their cavities to form a self-inclusion complex.19d On the other hand, tetraamine 5a adopts a 1,3-alternate conformation in its guest-free crystal.4d It is reported that this is because the circular hydrogen-bond network among the amino groups in compound 5a with a cone conformation is so weak as to be overcome by packing forces.20 In agreement with this, we recently found that compound 5a flexibly changes its conformation depending on the structure of guest molecules on forming inclusion crystals.13 Thus, tetraol 2 and tetraamine 5a exhibit different inclusion properties owing to the difference in strength of the circular hydrogen-bond network. As N,O-hybrid thiacalixarenes 5b and 5c were in hand, we examined the influence of replacing two hydroxy groups of tetraol 2 with amino groups on the strength of the circular hydrogen-bond network by X-ray crystallography.
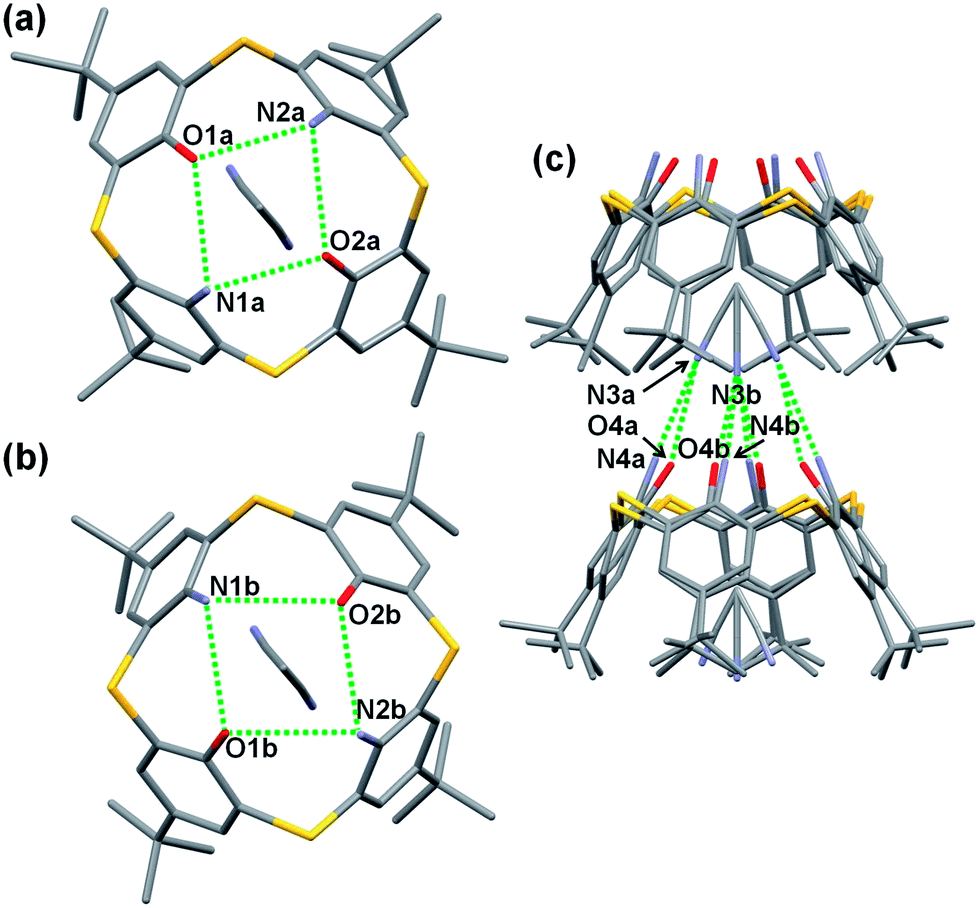
Single crystals of acetonitrile inclusion complexes formulated as 5b·CH3CN and 5c·CH3CN were obtained by slow diffusion of acetonitrile vapor into chloroform solutions of diamines 5b and 5c, respectively. X-ray analysis of these crystals revealed that they have similar packing structures to each other (Fig. 2 and 3). In each crystal, an acetonitrile molecule with disordered structures is included into the cavity of a calixarene with a cone conformation, directing the methyl group inside. The average distance between the methyl carbon and the centroid of each benzene ring for 5b·CH3CN is 3.623 Å and that for 5c·CH3CN is 3.647 Å, indicating the presence of a CH–π interaction. Intermolecular hydrogen bonds are observed between the cyano group of an acetonitrile molecule and the narrow rim substituents of an adjacent calixarene. Through the CH–π interaction and hydrogen bonds, an acetonitrile molecule joins two neighboring host molecules in a head-to-tail manner to construct an infinite columnar structure along the c-axis. A similar columnar structure mediated by acetonitrile molecules has also been found in inclusion crystals 2·CH3CN (ref. 19c) and 5a·CH3CN (ref. 13). However, the manners of the lamination of inclusion complexes are slightly different between 2·CH3CN and 5a–c·CH3CN; in 2·CH3CN, inclusion complexes of the same structure simply pile up, whereas a couple of complexes, a complex and its mirror image with regard to the a–c plane, pile up in 5a–c·CH3CN (Fig. 2c and 3b).13 This, combined with the fact that acetonitrile molecules are disordered in 5a–c·CH3CN but not in 2·CH3CN, suggests that not all of the hydrogen bonds between narrow rim substituents and an acetonitrile molecule have the same strength in each of inclusion crystals 5a–c·CH3CN. However, it could not be specified with which substituent an acetonitrile molecule strongly connects through a hydrogen bond, because the narrow rim substituents are disordered in 5a–c·CH3CN (Fig. S2 and S3†).13
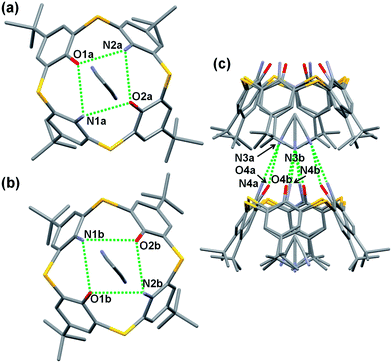 |
| | Fig. 2 X-ray structure of 5b·CH3CN. (a and b) Top views of disordered frameworks and (c) partial packing structure. Protons of the NH2 and OH groups were not found. Hydrogen atoms are omitted for clarity. Green dotted lines represent hydrogen bonds. Selected distances: N1a⋯O1a (3.057 Å), N1a⋯O2a (2.865 Å), O1b⋯N1b (3.114 Å), O1b⋯N2b (3.155 Å), N3a⋯N4a (3.156 Å), N3a⋯O4a (3.157 Å), N3b⋯N4b (3.386 Å), N3b⋯O4b (3.319 Å). | |
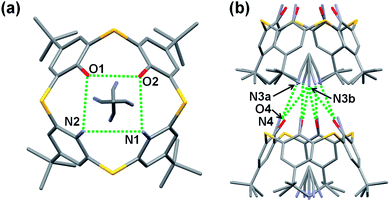 |
| | Fig. 3 X-ray structure of 5c·CH3CN. (a) Top view and (b) partial packing structure. Protons of the NH2 and OH groups were not found. Hydrogen atoms are omitted for clarity. Green dotted lines represent hydrogen bonds. Selected distances: O1⋯O2 (2.850 Å), O2⋯N1 (3.072 Å), N1⋯N2 (3.292 Å), N2⋯O1 (3.103 Å), N3a⋯N4 (3.155 Å), N3a⋯O4 (3.222 Å), N3b⋯N4 (3.404 Å), N3b⋯O4 (3.381 Å). | |
The relative strength of intramolecular hydrogen bonds between narrow rim substituents was estimated from interatomic distances between their N and/or O atoms for 5b·CH3CN and 5c·CH3CN. The mean distance between adjacent O and N atoms in 5b·CH3CN is 3.032 Å, which is longer than that between O atoms in 2·CH3CN (2.889 Å)19c and shorter than that between N atoms in 5a·CH3CN (3.298 Å).13 The O⋯O (2.850 Å), N⋯N (3.292 Å) and O⋯N distances (an average of 3.088 Å) in 5c·CH3CN are in reasonable agreement with the corresponding interatomic distances in 2·CH3CN (2.889 Å), 5a·CH3CN (3.298 Å) and 5b·CH3CN (3.032 Å), respectively. These observations indicate that the strength of the intramolecular hydrogen bonds falls in the order of O⋯O > O⋯N > N⋯N and that each kind of the hydrogen bonds has similar strength among the four acetonitrile complexes, regardless of the host molecules.
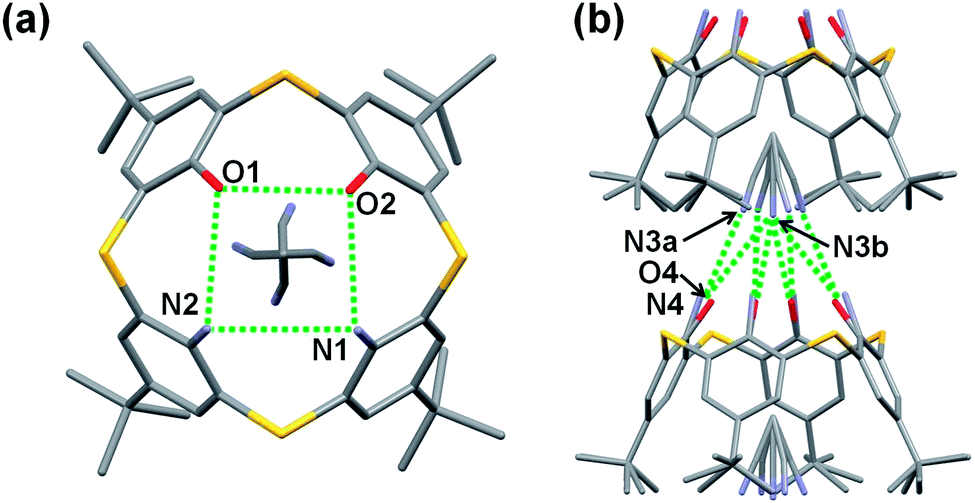
The question now arises whether the O⋯N hydrogen bonds are strong enough to stabilize the cone conformations of hybrid-type thiacalixarenes 5b and 5c in the crystals in the absence of guest molecules, such as acetonitrile. Then, compounds 5b and 5c were crystallized from various solvents in order to obtain guest-free single crystals. Slow diffusion of hexane vapor to a chloroform solution of 1,2-diamine 5c gave a desired crystal suitable for X-ray analysis (Fig. 4), while any attempts failed for 1,3-diamine 5b. In the crystal, compound 5c adopts a pinched cone conformation. Out of two pairs of facing benzene rings, one pair with a smaller dihedral angle is disordered (Fig. S4†); the ratio of the occupancies of the major and minor components is 7:3. Fig. 4 shows the major components. The O⋯O, N⋯N and O⋯N distances for the major components are 3.096 Å, 3.545 Å and an average of 3.197 Å, respectively, while those for the minor components are 3.347 Å, 3.151 Å and an average of 3.269 Å, respectively. These distances are different from those between the corresponding heteroatoms for the acetonitrile inclusion complex 5c·CH3CN (vide supra), but within the range of hydrogen bond lengths. The intermolecular O2⋯N2 distances between two neighboring calixarenes are 3.019 Å and 3.048 Å for the major and minor components, respectively, which indicates the presence of an intermolecular hydrogen bond. Through the intermolecular hydrogen bonds, a 21 helical structure is created along the b-axis (Fig. 4b). As mentioned earlier, tetraamine 5a adopts a 1,3-alternate conformation in its guest-free crystal.4d Since 1,2-diamine 5c adopts a cone conformation despite bearing an N,N-arrangement at the narrow-rim substituents, it can be said that the intramolecular O⋯N hydrogen bonds, not to mention the O⋯O hydrogen bond, play a crucial role to maintain the circular hydrogen-bond network in the crystal. It seems that these hybrid-type thiacalixarenes bear conformational characteristics similar to those of tetraol 2 rather than tetraamine 5a.
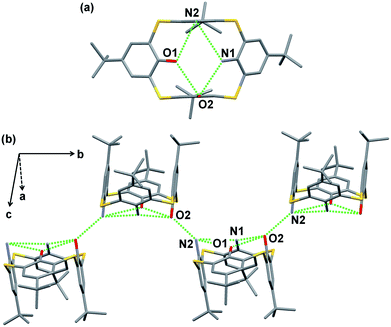 |
| | Fig. 4 X-ray structure of 5c. (a) Top view and (b) partial packing structure. Major components of disordered frameworks are shown. Hydrogen atoms of the NH2 and OH groups were not found. Hydrogen atoms are omitted for clarity. Selected distances for major components: O1⋯O2 (3.096 Å), O2⋯N1 (3.038 Å), O1⋯N2 (3.355 Å), N1⋯N2 (3.545 Å), N2⋯O2 (3.019 Å). | |
Conclusions
In this paper combined with a previous one,4d we have established a library synthesis of aminothiacalixarenes 5a–c via the benzylamination of tetra-O-methylsulfinylcalixarenes 4 with lithium benzylamide. The benzylamination of compounds 4 proceeded with perfect regioselectivity at the methoxy groups attached to the trans-benzene units, which was rationalized by the chelation-assisted SNAr mechanism. It is noteworthy that 1,2-diamine 5c is the first hybrid-type calix[4]arene with two substituents at the proximal positions. The X-ray crystallographic analysis of diamines 5b and 5c revealed that the introduction of amino groups into the calixarene framework instead of hydroxy groups weakens the circular hydrogen-bond network among the narrow-rim substituents. These compounds are attractive molecular scaffolds for the elaboration of sophisticated metal complexes because both soft amino and hard hydroxy groups are directly substituted at the narrow rim of a calixarene framework. In addition, compounds 2 and 5a–c are a series of host molecules, which have similar steric structures but are expected to show different conformational behaviours, depending on the strength of the circular hydrogen-bond network. Studies on the effect of this nature on molecular recognition ability are underway.13
Experimental
General
Mps were taken using a Stuart SMP3 apparatus and are uncorrected. Microanalyses were carried out in the Microanalytical Laboratory of the Institute of Multidisciplinary Research for Advanced Materials, Tohoku University. IR spectra were recorded on a Shimazu FTIR-8300 spectrometer. 1H and 13C NMR spectra were recorded on a Bruker DPX-400, DRX-500 or AV-400 spectrometer at 296 K, using tetramethylsilane as an internal standard and CDCl3 as a solvent, unless otherwise noted. J-values are given in Hz. FAB-MS were recorded on a JEOL GC-mate. HRMS spectra were measured using a Bruker Daltonics APEX III in Research and Analytical Center for Giant Molecules, Graduate School of Science, Tohoku University. Silica gel columns were prepared by use of Kanto Chemical silica gel 60N (63–210 μm). Water- and air-sensitive reactions were routinely carried out under nitrogen. Benzene was distilled from sodium diphenyl ketyl. Dry THF was purchased from Kanto Chemical. Compounds 4 were prepared as described previously.15
Syntheses
25,27-Di(benzylamino)-26,28-dimethoxy-tetra-p-tert-butylsulfinylcalix[4]arene (7b). To a solution of benzylamine (d 0.981; 2.0 cm3, 18 mmol) in dry THF (35 cm3) was added dropwise butyllithium (2.44 mol dm−3 in hexane; 3.5 cm3, 8.5 mmol) at −78 °C, and the mixture was stirred at 0 °C for 1.5 h. The mixture was added to a stirred suspension of compound 4(rctt) (923 mg, 1.10 mmol) in dry THF (80 cm3) in one portion at room temperature and stirring was continued for 5 min. After the reaction was quenched by the addition of saturated aqueous NH4Cl (10 cm3), the mixture was acidified with 2 M HCl (10 cm3), diluted with water (50 cm3) and extracted with chloroform (100 cm3 × 3). The combined organic layer was evaporated to leave a residue, which was purified by column chromatography with chloroform–ethyl acetate (20:1) as an eluent to give 1,3-dibenzylamine 7b (519 mg, 48%) as a colorless powder, mp 342–343 °C (found: C, 67.6; H, 6.8; N, 2.8%. Calc. for C56H66N2O6S4: C, 67.8; H, 6.7; N, 2.8%); νmax(KBr)/cm−1 3324 (OH), 2957 (CH), 1261 (CO), 1032 (SO) and 986 (CO); δH (400 MHz) 1.00 [9H, s, C(CH3)3], 1.06 [18H, s, C(CH3)3 × 2], 1.44 [9H, s, C(CH3)3], 3.30 (3H, s, OCH3), 3.85 (3H, s, OCH3), 4.60 (2H, dd, J 12.7 and 5.66, NHCH2 × 2), 4.66 (2H, dd, J 12.7 and 5.66, NHCH2 × 2), 6.17 (2H, t, J 5.66, NHCH2 × 2), 7.33–7.45 (14H, m, ArH), 7.83 (2H, s, ArH) and 8.15 (2H, s, ArH); δC (100 MHz) 30.72, 30.84, 31.42, 34.06, 34.97, 35.62, 54.34, 61.03, 61.12, 123.12, 125.00, 125.37, 128.03, 128.27, 128.99, 129.25, 130.70, 131.38, 134.73, 137.67, 138.51, 140.97, 145.62, 147.40, 147.90, 149.03 and 149.88; m/z (FAB) 991 [M + H+].
25,26-Di(benzylamino)-27,28-dimethoxy-tetra-p-tert-butylsulfinylcalix[4]arene (7c). This compound was prepared by the same procedure as used for the preparation of compound 7b. A solution of lithium benzylamide, prepared from benzylamine (5.5 cm3, 50 mmol) and butyllithium (1.56 mol dm−3 in hexane; 15.5 cm3, 24.2 mmol) in THF (40 cm3), was allowed to react with a solution of compound 4(rcct) (2.57 g, 3.06 mmol) in THF (100 cm3) for 8 min at room temperature. After the aqueous work-up, the crude product was purified by column chromatography with hexane–ethyl acetate (3:2) as an eluent to give 1,2-dibenzylamine 7c (948 mg, 31%) as a colorless powder, mp 159–161 °C; νmax(KBr)/cm−1 3448 (NH), 2963 (CH) and 1047 (SO); δH (400 MHz) 0.59 [9H, s, C(CH3)3], 1.05 [9H, s, C(CH3)3], 1.25 [9H, s, C(CH3)3], 1.41 [9H, s, C(CH3)3], 3.49 (3H, s, OCH3), 3.54–3.57 (1H, m, ArNHCH2), 4.08 (3H, s, OCH3), 4.29 (1H, t, J 10.1 Hz, NHCH2), 4.34–4.40 (1H, m, NHCH2), 4.68–4.70 (2H, m, NHCH2), 6.65 (1H, br, NHCH2), 6.93 (1H, d, J 2.3, ArH), 7.29–7.43 (8H, m, ArH), 7.48–7.50 (2H, m, ArH), 7.58–7.60 (2H, m, ArH), 7.81 (1H, d, J 2.3 Hz, ArH), 7.86 (1H, d, J 2.4, ArH), 8.03 (1H, d, J 2.3, ArH), 8.05 (1H, d, J 2.4, ArH) and 8.12 (1H, d, J 2.4, ArH); δC (100 MHz) 30.51, 30.75, 31.01, 31.37, 33.56, 34.57, 35.19, 35.66, 54.54, 56.15, 61.32, 63.29, 122.30, 123.83, 123.91, 124.34, 125.04, 125.67, 127.84, 127.91, 128.26, 128.87, 128.94, 129.11, 129.41, 130.00, 130.67, 131.90, 133.89, 136.01, 136.05, 137.65, 137.78, 138.41, 138.83, 139.25, 139.94, 144.45, 144.54, 146.25, 148.13, 149.66, 149.71 and 151.48; m/z (FAB) 991 [M + H+]; HRMS (ESI): calc. for C56H66N2NaO6S4 [M + Na+] 1013.3696, found 1013.3691.
4-tert-Butyl-2-(butylsulfinyl)-1-methoxybenzene (8). To a suspension of compound 4(rtct) (131 mg, 0.156 mmol) in dry THF (2.0 cm3) was added dropwise butyllithium (1.57 mol dm−3 in hexane, 1.6 cm3, 2.5 mmol) at −78 °C, and the mixture was stirred at 0 °C for 1 h. After the reaction was quenched with aqueous NH4Cl, the mixture was acidified with 2 M HCl, diluted with water and extracted with chloroform. The extracts were evaporated to leave a residue, which was purified by thin layer chromatography with chloroform–ethyl acetate (50:1) as an eluent to give compound 8 (102 mg, 61%) as a colorless oil, νmax(chloroform solution)/cm−1 1223 (CO), 1030 (SO) and 907 (CO); δH (400 MHz) 0.93 (3H, t, J 7.3 Hz, CH2CH2CH2CH3), 1.34 [9H, s, C(CH3)3], 1.39–1.52 (2H, m, CH2CH2CH2CH3), 1.58–1.67 (1H, m, CH2CH2CH2CH3), 1.80–1.91 (1H, m, CH2CH2CH2CH3), 2.69–2.76 (1H, m, CH2CH2CH2CH3), 3.01–3.08 (1H, m, CH2CH2CH2CH3), 3.86 (3H, s, OCH3), 6.85 (1H, d, J 8.6 Hz, ArH), 7.43 (1H, dd, J 8.6 and 2.4 Hz, ArH) and 7.80 (1H, d, J 2.4 Hz, ArH); δC (125 MHz) 13.77, 21.98, 24.39, 31.52, 34.74, 53.84, 55.83, 110.30, 122.19, 128.51, 130.59, 144.87 and 152.84; HRMS (EI): calc. for C15H24O2S 268.1497, found 268.1494.
25,27-Diamino-26,28-dimethoxy-tetra-p-tert-butylsulfinylcalix[4]arene (9b). To a solution of 1,3-dibenzylamine 7b (1.14 g, 1.15 mmol) and NBS (1.23 g, 6.91 mmol) in dry benzene (100 cm3) was added BPO (134 mg, 0.55 mmol) at room temperature, and the mixture was refluxed for 1.5 h. After cooling, the reaction was quenched with 2 M HCl (30 cm3) and the two layers were separated. The aqueous layer was extracted with chloroform and the combined organic layer was evaporated to dryness. The residue was dissolved in chloroform (40 cm3) and boiled with 6 M HCl (40 cm3) for 12 h. After cooling, the two layers were separated and the aqueous layer was extracted with chloroform. The combined extracts were evaporated to leave a residue, which was purified by column chromatography with chloroform–ethyl acetate (2:1) as an eluent to give amino ether 9b (738 mg, 90%) as a colorless powder, mp 310–311 °C (decomp.); νmax(KBr)/cm−1 3343, 3337 (NH), 1269 (CO), 1039 (SO) and 991 (CO); δH (500 MHz) 1.03 [18H, s, C(CH3)3 × 2], 1.45 [18H, s, C(CH3)3 × 2], 3.72 (6H, s, OCH3 × 2), 5.23 (4H, s, NH2 × 2), 7.24 (4H, s, ArH × 2) and 8.16 (4H, s, ArH × 2); δC (125 MHz) 30.84, 31.34, 33.99, 35.51, 61.53, 124.72, 126.66, 128.62, 135.93, 140.52, 143.71, 148.17 and 148.65; m/z (FAB) 811 [M + H+]; HRMS (ESI): calc. for C42H54N2NaO6S4 [M + Na+] 833.2757, found 833.2753.
25,26-Diamino-27,28-dimethoxy-tetra-p-tert-butylsulfinylcalix[4]arene (9c). This compound was prepared by the same procedure as mentioned for the preparation of compound 9b. A mixture of 1,2-dibenzylamine 7c (963 mg, 0.971 mmol), NBS (1.04 g, 5.84 mmol), BPO (113 mg, 0.47 mmol) and benzene (60 cm3) was refluxed for 2 h. The crude product was dissolved in chloroform (30 cm3) and boiled with 6 M HCl (30 cm3) for 2 h to give, after chromatographic purification with chloroform–ethyl acetate (6:1) as an eluent, amino ether 9c (502 mg, 64%) as a colorless powder, mp 310.9–312.6 °C (decomp.) (found: C, 61.89; H, 6.76; N, 3.49%. Calc. for C42H54N2O6S4: C, 62.19; H, 6.71; N, 3.45); νmax(KBr)/cm−1 3448, 3335 (NH), 2961 (CH) and 1040 (SO); δH (400 MHz, [2H2]-1,1,2,2-tetrachloroethane, 383 K): 1.18 [18H, s, C(CH3)3 × 2], 1.25 [18H, s, C(CH3)3 × 2], 3.90 (6H, s, OCH3 × 2), 5.01 (4H, s, NH2 × 2), 7.45 (2H, d, J 2.2 Hz, ArH), 7.52 (2H, br s, ArH), 7.75 (2H, d, J 2.2 Hz, ArH) and 7.91 (2H, d, J 2.2 Hz, ArH); δC (100 MHz, [2H2]-1,1,2,2-tetrachloroethane, 383 K): 31.13, 31.18, 34.23, 35.39, 62.99, 124.53, 124.65, 125.15, 127.24, 127.58, 128.80, 137.41, 138.92, 140.38, 142.86, 149.79 and 150.02; m/z (FAB) 811 [M + H+].
25,27-Diamino-26,28-dihydroxy-tetra-p-tert-butylsulfinylcalix[4]arene (10b). To a solution of 1-octanethiol (d 0.843; 2.0 cm3, 12 mmol) in dry THF (30 cm3) was added NaH (60 wt% in mineral oil; 315 mg, 7.68 mmol) at 0 °C, and the mixture was stirred at 50 °C for 1 h. To the suspension was added amino ether 9b (764 mg, 0.942 mmol), and the mixture was refluxed for 18 h. After cooling, the mixture was acidified with 2 M HCl (20 cm3) and the resulting mixture was extracted with chloroform. The organic layer was evaporated to leave a residue, which was crystallized from methanol to give diol 10b (639 mg, 87%) as a colorless powder, mp 216–217 °C (decomp.); νmax(KBr)/cm−1 3450, 3362 (NH), 3224 (OH), 2959 (CH) and 1040 (SO); δH (400 MHz, [2H6]DMSO, 353 K) 1.05 [18H, s, C(CH3)3 × 2], 1.34 [18H, s, C(CH3)3 × 2], 7.37 (4H, s, ArH × 2) and 7.84 (4H, s, ArH × 2); δC (100 MHz, [2H6]DMSO, 353 K) 30.24, 30.62, 33.27, 34.13, 122.93, 126.64, 127.66, 131.73, 138.55, 141.93, 142.90, 142.90 and 147.05; m/z (FAB) 783 [M + H+]; HRMS (ESI): calc. for C40H50N2NaO6S4 [M + Na+] 805.2444, found 805.2441.
25,26-Diamino-27,28-dihydroxy-tetra-p-tert-butylsulfinylcalix[4]arene (10c). This compound was prepared by the same procedure as mentioned for the preparation of compound 10b. Sodium octanethiolate prepared from 1-octanethiol (520 mm3, 3.00 mmol) and NaH (60 wt% in mineral oil, 79 mg, 1.9 mmol) in THF (20 cm3) was refluxed with amino ether 9c (200 mg, 0.247 mmol) for 5 h. After the work-up, the crude product was crystallized from methanol to give diol 10c (175 mg, 90%) as a colorless powder, mp 228 °C (decomp.); νmax(KBr)/cm−1 3341 (NH), 3215 (OH) and 1038 (SO); δH (400 MHz, [2H6]DMSO, 373 K) 1.08 [18H, s, C(CH3)3 × 2], 1.25 [18H, s, C(CH3)3 × 2], 6.72 (6H, br, NH2 × 2 and OH × 2), 7.34 (2H, br, ArH), 7.46 (2H, d, J 2.3 Hz, ArH), 7.56 (2H, d, J 2.5 Hz, ArH) and 7.71 (2H, d, J 2.5 Hz, ArH); δC (100 MHz, [2H6]DMSO, 373 K) 30.11, 30.53, 33.34, 33.83, 121.64, 121.86, 124.29, 125.26, 126.27, 132.34, 132.70, 139.08, 140.77, 142.77, 142.98, 145.80 and 151.21; m/z (FAB) 783 [M + H+]; HRMS (ESI): calc. for C40H50N2NaO6S4 [M + Na+] 805.2444, found 805.2443.
25,27-Diamino-26,28-dihydroxy-tetra-p-tert-butythiacalix[4]arene (5b). To a mixture of diol 10b (80.3 mg, 0.103 mmol), LiAlH4 (64.0 mg, 1.69 mmol) and dry THF (16 cm3) was added dropwise TiCl4 (d 1.726; 90 mm3, 0.82 mmol) at −78 °C, and the mixture was stirred at room temperature for 4 h. After the reaction was quenched by the addition of saturated aqueous NH4Cl (10 cm3) at 0 °C, the mixture was acidified with 2 M HCl (10 cm3) and extracted with chloroform. The organic layer was evaporated to leave a residue, which was purified by column chromatography with chloroform–ethyl acetate (30:1) as an eluent to give 1,3-diaminothiacalixarene 5b (54 mg, 73%) as a colorless powder, mp 290–292 °C (decomp); νmax(KBr)/cm−1 3445, 3395 (NH), 3356 (OH) and 2963 (CH); δH (400 MHz) 1.13 [18H, s, C(CH3)3 × 2], 1.25 [18H, s, C(CH3)3 × 2], 7.49 (4H, s, ArH × 2) and 7.61 (4H, s, ArH × 2); δC (100 MHz) 31.05, 31.39, 34.04, 34.14, 121.60, 123.89, 134.79, 135.41, 142.85, 145.31, 146.42 and 157.21; m/z (FAB) 719 [M + H+]; HRMS (ESI) calc. for C40H50N2NaO2S4 [M + Na+] 741.2647, found 741.2644.
25,26-Diamino-27,28-dihydroxy-tetra-p-tert-butythiacalix[4]arene (5c). This compound was prepared by a similar procedure to that used for the preparation of compound 5b. A mixture of 10c (160 mg, 0.204 mmol), LiAlH4 (123 mg, 3.24 mmol), TiCl4 (175 mm3, 1.59 mmol) and THF (20 cm3) was stirred at room temperature for 3 h as mentioned above. After the work-up, the crude product was dissolved in THF (10 cm3). To the mixture was added TBAF (1.0 mol dm−3 in THF; 200 mm3, 0.20 mmol), and the resulting mixture was stirred at room temperature for 3 h. After the addition of water (40 cm3), the mixture was extracted with chloroform and the extracts were evaporated to dryness. The residue was purified by column chromatography with chloroform as an eluent to give 1,2-diaminothiacalixarene 5c (68 mg, 46%) as a colorless powder, mp 331–333 °C (decomp.) (found: C, 66.7; H, 6.9; N, 3.9%. Calc. for C40H50N2O2S4: C, 66.8; H, 7.0; N, 3.9%); νmax(KBr)/cm−1 3464, 3418 (NH), 3356 (OH) and 2963 (CH); δH (400 MHz) 1.14 [18H, s, C(CH3)3 × 2], 1.20 [18H, s, C(CH3)3 × 2], 6.32 (6H, br, NH2 × 2 and OH × 2), 7.39 (2H, d, J 2.3 Hz, ArH), 7.47 (2H, d, J 2.3 Hz, ArH), 7.49 (2H, d, J 2.5 Hz, ArH) and 7.54 (2H, d, J 2.5 Hz, ArH); δC (100 MHz) 31.16, 31.24, 33.95, 34.09, 119.75, 120.12, 120.61, 120.99, 134.15, 134.23, 134.42, 134.77, 142.58, 143.69, 146.70 and 155.38; m/z (FAB) 719 [M + H+].
X-ray crystallography
Diffraction data of 7c·CH3OH·H2O, 5b·CH3CN and 5c were collected with a Rigaku MSC Mercury CCD diffractometer, using Mo-Kα radiation (λ = 0.71069 Å). The structures were solved by the direct method and refined by full-matrix least-square calculations, using TeXsan (v. 1.11).21 Diffraction data of 5c·CH3CN were collected with a Bruker APEX II CCD diffractometer equipped with a Helios multi-layered confocal mirror and a TXS fine-focus rotating anode, using Mo-Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined by full-matrix least-squares on F2 using SHELXS and SHELXL.22 All materials for publication were prepared by Yadokari-XG 2009 software.†,23
Data for 7c·CH3OH·H2O. C57H72N2O8S4, M = 1041.41, monoclinic, P21/c, a = 13.8234(9) Å, b = 19.7567(10) Å, c = 24.5819(13) Å, α = 90°, β = 123.419(3)°, γ = 90°, V = 5603.5(5) Å3, Z = 4, Dcalc = 1.234 g cm−3, μ(Mo-Kα) = 0.223 mm−1, T = 113 K, 44910 reflections measured, 9701 independent reflections (Rint = 0.0328), 7721 reflections were observed (I > 2σ(I)), R1 = 0.0635, wR2 = 0.1695 (observed), R1 = 0.0833, wR2 = 0.1811 (all data), GOF = 1.324.
Data for 5b·CH3CN. C42H53N3O2S4, M = 757.09, orthorhombic, Pccn, a = 15.400(5) Å, b = 15.405(5) Å, c = 17.403(5) Å, α = 90°, β = 90°, γ = 90°, V = 4129(2) Å3, Z = 4, Dcalc = 1.218 g cm−3, μ(Mo-Kα) = 0.268 mm−1, T = 113 K, 30747 reflections measured, 30747 independent reflections (Rint = 0.0000), 21020 reflections were observed (I > 2σ(I)), R1 = 0.0974, wR2 = 0.2057 (observed), R1 = 0.1383, wR2 = 0.2325 (all data), GOF = 1.066. The positions of disordered acetonitrile molecule were isotropically refined without any restraints at two positions with same site occupancy. The positions of calixarene frameworks were anisotropically refined at two positions with same site occupancy. All disordered benzene rings were restrained using FLAT and SAME restraints so that the benzene rings are planar and have symmetry. Likewise, the disordered tert-butyl groups were restrained using SAME restraints. In addition, SIMU restraints were used owing to the high degree of disorder in this structure. All hydrogen atoms placed in calculated positions and refined as riding atoms.
Data for 5c·CH3CN. C42H53N3O2S4, M = 760.11, tetragonal, P4/ncc, a = 15.3887(16) Å, b = 15.3887(16) Å, c = 17.563(3) Å, α = 90°, β = 90°, γ = 90°, V = 4159.2(9) Å3, Z = 4, Dcalc = 1.214 g cm−3, μ(Mo-Kα) = 0.272 mm−1, T = 173 K, 21688 reflections measured, 2396 independent reflections (Rint = 0.0937), 1447 reflections were observed (I > 2σ(I)), R1 = 0.0769, wR2 = 0.2036 (observed), R1 = 0.1247, wR2 = 0.2528 (all data), GOF = 1.102. The positions of disordered acetonitrile molecule were isotropically refined without any restraints at two positions with same site occupancy. The positions of disordered tert-butyl group were anisotropically refined at two positions with same site occupancy. The benzene ring was restrained using FLAT restraints. The disordered tert-butyl groups were restrained using SAME restraints. In addition, SIMU restraints were used owing to the high degree of disorder in this structure. The position of disordered acetonitrile molecule was separated into two parts with the equal occupancies, and isotropically refined without any restraints. All hydrogen atoms placed in calculated positions and refined as riding atoms.
Data for 5c. C40H50N2O2S4, M = 719.06, monoclinic, P21, a = 10.019(5) Å, b = 15.041(7) Å, c = 13.613(7) Å, α = 90°, β = 103.487(13)°, γ = 90°, V = 1994.9(17) Å3, Z = 2, Dcalc = 1.197 g cm−3, μ(Mo-Kα) = 0.273 mm−1, T = 113 K, 14753 reflections measured, 7564 independent reflections (Rint = 0.0604), 5684 reflections were observed (I > 2σ(I)), R1 = 0.0903, wR2 = 0.2370 (observed), R1 = 0.1132, wR2 = 0.2547 (all data), GOF = 1.122. The positions of disordered tert-butyl groups and calixarene frameworks were isotropically refined at two positions with optimal occupancy. All disordered benzene rings were restrained using FLAT and SAME restraints so that the benzene rings are planar and have symmetry. Likewise, the disordered tert-butyl groups were restrained using SAME restraints. In addition, SIMU restraints were used owing to the high degree of disorder in this structure. All hydrogen atoms placed in calculated positions and refined as riding atoms.
Notes and references
- Reviews:
(a) C. D. Gutsche, Calixarenes, Monographs in Supramolecular Chemistry, ed. J. F. Stoddart, The Royal Society of Chemistry, Cambridge, 1989 Search PubMed;
(b) V. Böhmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 713–745 CrossRef;
(c) A. Ikeda and S. Shinkai, Chem. Rev., 1997, 97, 1713–1734 CrossRef CAS PubMed;
(d) C. D. Gutsche, Calixarenes revisited, Monographs in Supramolecular Chemistry, ed. J. F. Stoddart, The Royal Society, Cambridge, 1998 Search PubMed;
(e) L. Mandolini and R. Ungaro, Calixarenes in Action, Imperial College Press, London, 2000 Search PubMed;
(f) Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Calixarenes 2001, Kluwer Academic, Dordrecht, 2001 Search PubMed.
- Reviews:
(a) N. Iki and S. Miyano, J. Inclusion Phenom. Macrocyclic Chem., 2001, 41, 99–105 CrossRef CAS;
(b) P. Lhoták, Eur. J. Org. Chem., 2004, 1675–1692 CrossRef;
(c) S. Parola and C. Desroches, Collect. Czech. Chem. Commun., 2004, 69, 966–983 CrossRef CAS;
(d) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291–5316 CrossRef CAS PubMed.
- Replacement with mercapto groups:
(a) C. G. Gibbs and C. D. Gutsche, J. Org. Chem., 1993, 3, 5338–5339 Search PubMed;
(b) C. G. Gibbs, P. K. Sujeeth, J. S. Rogers, G. G. Stanley, M. Krawiec, W. H. Watson and C. D. Gutsche, J. Org. Chem., 1995, 60, 8394–8402 CrossRef CAS;
(c) P. Rao, M. W. Hosseini, A. D. Cian and J. Fischer, Chem. Commun., 1999, 2169–2170 RSC;
(d) R. Zieba, C. Desroches, F. Chaput, C. Sigala, E. Jeanneau and S. Parola, Tetrahedron Lett., 2007, 48, 5401–5405 CrossRef CAS.
- Replacement with amino groups:
(a) F. Ohseto, H. Murakami, K. Araki and S. Shinkai, Tetrahedron Lett., 1992, 33, 1217–1220 CrossRef CAS;
(b) O. Aleksiuk, F. Grynszpan and S. E. Biali, J. Org. Chem., 1993, 58, 1994–1996 CrossRef CAS;
(c) O. Aleksiuk, S. Cohen and S. E. Biali, J. Am. Chem. Soc., 1995, 117, 9645–9652 CrossRef CAS;
(d) H. Katagiri, N. Iki, T. Hattori, C. Kabuto and S. Miyano, J. Am. Chem. Soc., 2001, 123, 779–780 CrossRef CAS PubMed;
(e) Y. Nakamura, S. Tanaka, R. Serizawa, N. Morohashi and T. Hattori, J. Org. Chem., 2011, 76, 2168–2179 CrossRef CAS PubMed.
- Replacement with alkynyl groups: H. Al-Saraierh, D. O. Miller and P. E. Georghiou, J. Org. Chem., 2005, 70, 8273–8280 CrossRef CAS PubMed.
- Replacement with halo groups: J. M. Van Gelder, O. Aleksiuk and S. E. Biali, J. Org. Chem., 1996, 61, 8419–8424 CrossRef CAS ; ref. 5 and 14.
- Replacement with phosphono groups: N. Morohashi, T. Hayashi, Y. Nakamura, T. Kobayashi, S. Tanaka and T. Hattori, Chem. Lett., 2012, 41, 1520–1522 CrossRef CAS.
-
(a) T. Hattori, T. Suzuki, N. Hayashizaka, N. Koike and S. Miyano, Bull. Chem. Soc. Jpn., 1993, 66, 3034–3040 CrossRef CAS;
(b) T. Hattori, J. Sakamoto, N. Hayashizaka and S. Miyano, Synthesis, 1994, 199–202 CrossRef CAS;
(c) T. Hattori, M. Suzuki, Y. Komuro and S. Miyano, J. Chem. Soc., Perkin Trans. 1, 1995, 1473–1474 RSC;
(d) T. Hattori, M. Suzuki, N. Tomita, A. Takeda and S. Miyano, J. Chem. Soc., Perkin Trans. 1, 1997, 1117–1123 RSC;
(e) T. Hattori, A. Takeda, K. Suzuki, N. Koike, E. Koshiishi and S. Miyano, J. Chem. Soc., Perkin Trans. 1, 1998, 3661–3671 RSC;
(f) T. Hattori, Y. Shimazumi, H. Goto, O. Yamabe, N. Morohashi, W. Kawai and S. Miyano, J. Org. Chem., 2003, 68, 2099–2108 CrossRef CAS PubMed.
- For recent progress of chelation-assisted SNAr reaction, see:
(a) J. S. Parker, N. A. Smith, M. J. Welham and W. O. Moss, Org. Process Res. Dev., 2004, 8, 45–50 CrossRef CAS;
(b) A. J. Brockway, M. González-López, J. C. Fettinger and J. T. Shaw, J. Org. Chem., 2011, 76, 3515–3518 CrossRef CAS PubMed;
(c) R. Aissaoui, A. Nourry, A. Coquel, T. T. H. Dao, A. Derdour, J.-J. Helesbeux, O. Duval, A.-S. Castanet and J. Mortier, J. Org. Chem., 2012, 77, 718–724 CrossRef CAS PubMed;
(d) G. Jiménez-Osés, A. J. Brockway, J. T. Shaw and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 6633–6642 CrossRef PubMed.
- Reviews for Meyers reaction:
(a) M. Reuman and A. I. Meyers, Tetrahedron, 1985, 41, 837–860 CrossRef CAS;
(b) T. G. Gant and A. I. Meyers, Tetrahedron, 1994, 50, 2297–2360 CrossRef CAS;
(c) J. Mortier, Curr. Org. Chem., 2011, 15, 2413–2437 CrossRef CAS.
- H. Katagiri, N. Iki, Y. Matsunaga, C. Kabuto and S. Miyano, Chem. Commun., 2002, 2080–2081 RSC.
-
(a) N. Iki, N. Morohashi, F. Narumi and S. Miyano, Bull. Chem. Soc. Jpn., 1998, 71, 1597–1603 CrossRef CAS;
(b) N. Morohashi, N. Iki, A. Sugawara and S. Miyano, Tetrahedron, 2001, 57, 5557–5563 CrossRef CAS.
- For inclusion properties of tetraamine 5a toward small organic molecules, see: N. Morohashi, H. Katagiri, T. Shimazaki, Y. Kitamoto, S. Tanaka, C. Kabuto, N. Iki, T. Hattori and S. Miyano, Supramol. Chem., 2013, 25, 812–818 CrossRef CAS.
- Part of this work has been published as a preliminary communication: S. Tanaka, H. Katagiri, N. Morohashi, T. Hattori and S. Miyano, Tetrahedron Lett., 2007, 48, 5293–5296 CrossRef CAS.
- N. Morohashi, H. Katagiri, N. Iki, Y. Yamane, C. Kabuto, T. Hattori and S. Miyano, J. Org. Chem., 2003, 68, 2324–2333 CrossRef CAS PubMed.
- H. Katagiri, T. Hattori, N. Morohashi, N. Iki and S. Miyano, J. Org. Chem., 2007, 72, 8327–8331 CrossRef CAS PubMed.
- See ESI for detail.†.
-
(a) O. Aieksiuk, F. Grynszpan and S. E. Biali, J. Chem. Soc., Chem. Commun., 1993, 11–13 RSC;
(b) J. Espinas, U. Darbost, J. Pelletier, E. Jeanneau, C. Duchamp, F. Bayard, O. Boyron, J.-P. Broyer, J. Thivolle-Cazat, J.-M. Basset, M. Taoufik and I. Bonnamour, Eur. J. Inorg. Chem., 2010, 9, 1349–1359 CrossRef.
-
(a) H. Akdas, L. Bringel, E. Graf, M. W. Hosseini, G. Mislin, J. Pansanel, A. De Cian and J. Fischer, Tetrahedron Lett., 1998, 39, 2311–2314 CrossRef CAS;
(b) N. Iki, C. Kabuto, T. Fukushima, H. Kumagai, H. Takeya, S. Miyanari, T. Miyashi and S. Miyano, Tetrahedron, 2000, 56, 1437–1443 CrossRef CAS;
(c) A. Arduini, F. F. Nachtigall, A. Pochini, A. Secchi and F. Ugozzoli, Supramol. Chem., 2000, 12, 273–291 CrossRef CAS;
(d) A. Bilyk, A. K. Hall, J. M. Harrowfield, M. W. Hosseini, B. W. Skelton and A. H. White, Inorg. Chem., 2001, 40, 672–686 CrossRef CAS PubMed;
(e) N. Morohashi, S. Noji, H. Nakayama, Y. Kudo, S. Tanaka, C. Kabuto and T. Hattori, Org. Lett., 2011, 13, 3292–3295 CrossRef CAS PubMed.
- E. E. Zvereva, S. A. Katsyuba, A. E. Vandyukov, A. V. Chernova, V. I. Kovalenko, S. E. Solovieva, I. S. Antipin and A. I. Konovalov, Spectrochim. Acta, Part A, 2010, 75, 872–879 CrossRef PubMed.
- TeXsan Single Crystal Structure Analysis Software, Molecular Structure Corporation, 3200 Research Forest Drive, The Woodlands, TX 77381, USA; Rigaku, 3-9-12 Akishima, Tokyo, Japan, 1999 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
-
(a) K. Wakita, Yadokari-XG: Software for crystal Structure Analyses, 2001 Search PubMed;
(b) C. Kabuto, S. Akine, T. Nemoto and E. Kwon, Yadokari-XG 2009: Release of Software for Crystal Structure Analyses, J. Crystallogr. Soc. Jpn., 2009, 51, 218–224 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional Scheme S1 and Fig. S1–S4. Copy of 1H and 13C NMR spectral charts for all new compounds. CCDC 974895–974898. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra47718e |
| ‡ Present address: Graduate School of Science and Engineering, Yamagata University, 4-3-16 Jonan, Yonezawa, Yamagata 992-8510, Japan. |
| § Present address: Department of Environmental Studies, Graduate School of Environmental Studies, Tohoku University, 6-6-07 Aramaki-Aoba, Aoba-ku, Sendai 980-8579, Japan. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.