
Flexible TiO2/cellulose acetate hybrid film as a recyclable photocatalyst†
Abstract
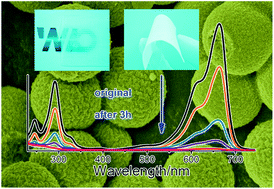
In this work, a flexible mesoporous TiO2 microspheres/cellulose acetate (TCA) hybrid film of tunable size and transparency was designed as a high performance recyclable photocatalyst. It was obtained by a simple method of dispersing mesoporous TiO2 microspheres onto the surface of a free-standing cellulose acetate (CA) film at room temperature. The photoelectrochemical properties of the mesoporous TiO2 microspheres were first studied by configuring them as a simple self-powered photoelectrochemical cell (PEC). Under UV light irradiation, the TCA hybrid film displays excellent flexibility and favorable recyclable photocatalytic activity for the decomposition of methylene blue (MB) solution. It was found that the pH value of the solution has a more significant effect than temperature on the photoactivity of the sample. Our results indicate that the hybrid film can be easily applied in the field of wastewater treatment without leaving any photocatalyst in the reaction system. It is feasible to develop this simple and environmentally friendly method to synthesize other catalyst systems such as WO3 film for potential industrial applications.
 Please wait while we load your content...
Please wait while we load your content...

