 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of functionalized α-trifluoroethyl amine scaffolds via Grignard addition to N-aryl hemiaminal ethers†
Amrei
Deutsch
a,
Herbert
Glas
b,
Anja
Hoffmann-Röder
*a and
Carl
Deutsch
*b
aCenter For Integrated Protein Science Munich (CIPSM) at the Department of Chemistry, Ludwig-Maximilians-Universitaet, Butenandtstr. 5–13, 81377 Munich, Germany. E-mail: Anja.Hoffmann-Roeder@cup.lmu.de; Fax: +49 89 2180 77914; Tel: +49 89 2180 77913
bMedicinal Chemistry, Merck, Frankfurter Strasse 250, 64293 Darmstadt, Germany. E-mail: Carl.Deutsch@merckgroup.com; Tel: +49 6151 72 2500
First published on 23rd January 2014
Abstract
The synthesis of a variety of α-branched trifluoroethyl amines was achieved by reaction of N-aryl hemiaminal ethers with organomagnesium reagents.
The design of compounds suitable for drug development is a multi-dimensional process that requires the fine tuning of molecular properties beyond potency. For instance, physical–chemical properties that directly affect hydrolytic stability and bioavailability of the compound must be addressed to avoid side effects in vivo. Due to the excellent pharmacological profile of fluorinated drugs, the strategic incorporation of fluorine atoms has nowadays become routine in medicinal chemistry development programs.1 For instance, incorporation of a trifluoromethyl substituent adjacent to an amine is a means to improve the metabolic stability and to attenuate the basicity of the compound by shifting its pKa value more towards those of amides.2 Furthermore the C–CF3 bond is substantially isopolar with the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and the trifluoroethyl amine moiety has a structural similarity to the tetrahedral proteolytic transition state.3 As a consequence, trifluoroethyl amines can be used as versatile hydrolysis-resistant bioisosteres of amides4 which retain the geometry of the amide bond and, in contrast to other mimetics5, also preserve the donating properties of the N–H bond. An illustrative example for the successful replacement of the amide functionality by a trifluoroethyl amine is given by Odanacatib, a highly potent drug candidate for the inhibition of Cathepsin K.6
O bond and the trifluoroethyl amine moiety has a structural similarity to the tetrahedral proteolytic transition state.3 As a consequence, trifluoroethyl amines can be used as versatile hydrolysis-resistant bioisosteres of amides4 which retain the geometry of the amide bond and, in contrast to other mimetics5, also preserve the donating properties of the N–H bond. An illustrative example for the successful replacement of the amide functionality by a trifluoroethyl amine is given by Odanacatib, a highly potent drug candidate for the inhibition of Cathepsin K.6
Various methods for the synthesis of α-trifluoromethylated amines have been described so far,7 including hydrogenation8 and aromatic substitution9 of activated imines, as well as base-catalyzed asymmetric isomerization reactions of ketoimines.10 Furthermore, nucleophilic addition reactions of various organometallics to trifluoromethylated imines11 and hydrazones12 have been reported. In this context, Lauzon and Charette have shown that trifluoromethyl amine derivatives can be prepared by copper-catalyzed nucleophilic addition of diorganozinc reagents to N-phosphinoylimines, using an excess of organozinc reagent.13 Similarly, trifluoromethylated α,α-dibranched carbinamines can be obtained from N-tert-butylsulfinyl hemiaminals with organomagnesium or organolithium reagents.14 However, most of these approaches are either hampered by the high tendency of α,α,α-trifluorethylimines to form hydrates or by the need of additional deprotection steps for further functionalization of the nitrogen atom. In a seminal paper, Mikami and coworkers showed that an excess of Grignard reagents can be used to prepare α-trifluoromethylated amines from stable N,O-acetals of trifluoroacetaldehyd.15,16 This work was recently extended to the use of arylboroxines for palladium(II)-catalyzed synthesis of α-(trifluoromethyl)arylmethyl amines.17
Herein, we report a systematic study on the synthesis of functionalized α-substituted trifluoromethyl amines using Grignard reagents and readily available trifluoromethylated hemiaminal ethers.18 The latter are shelf-stable compounds derived from 1-ethoxy-2,2,2-trifluoroethanol and aromatic amines and can be converted into trifluoromethylated aldimines in situ. Thus, upon treatment with Grignard reagent deprotonation should provide the corresponding imine which would then undergo nucleophilic attack by excess Grignard reagent to furnish the desired trifluoromethyl amine (Scheme 1). Formation of the transient imine species was confirmed by observing the corresponding imine hydrate via HPLC-MS after addition of MeMgBr to the reaction mixture.
 | ||
| Scheme 1 | ||
To determine the optimal conditions, 3-chloro-N-(1-ethoxy-2,2,2-trifluoroethyl)aniline 1a was treated with MeMgBr in dry THF under argon at different reaction temperatures (Table 1). Thus, the addition proceeded smoothly at −78 °C furnishing the desired trifluoroethyl amine 2a in 74% yield after one hour. Whereas higher temperatures above 0 °C led to significant formation of side and decomposition products, best yields were obtained at a temperature of −15 °C (Table 1, entries 1–5). For complete conversion of the trifluoromethyl N,O-acetals at least 2 eq. MeMgBr are required. However, larger excess of the nucleophile did not improve the yield significantly. Similar results were obtained with N-(1-ethoxy-2,2,2-trifluoroethyl)aniline 1b bearing a neutral aryl ring (Table 1, entries 7–9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | Eq. [MeMgBr] | T [°C] | Yield (%)a | Product |
| a Yield of isolated product after flash chromatography. b Reaction time of 2 h. | |||||
| 1 | 3-ClC6H4 | 2 | 40 | 65 | 2a |
| 2 | 3-ClC6H4 | 2 | 25 | 70 | 2a |
| 3 | 3-ClC6H4 | 2 | 0 | 84 | 2a |
| 4 | 3-ClC6H4 | 2 | −15 | 87 | 2a |
| 5 | 3-ClC6H4 | 2 | −78 | 74 | 2a |
| 6 | 3-ClC6H4 | 3 | −15 | 75 | 2a |
| 7 | C6H5 | 1 | −15 | 34b | 2b |
| 8 | C6H5 | 2 | −15 | 63 | 2b |
| 9 | C6H5 | 3 | −15 | 65 | 2b |
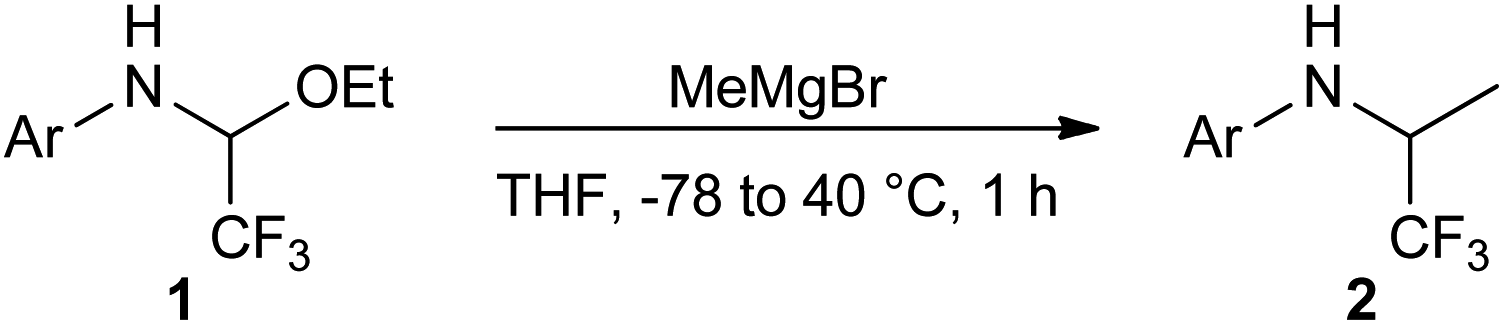
With the optimized reaction conditions in hands, the substrate scope of the nucleophilic addition was tested using various functionalized aryl N,O-acetals 1 and MeMgBr (Table 2). We were pleased to find that besides halides also ester, triazole, trifluoromethyl groups and morpholino substituents are well tolerated to provide the desired trifluoroethyl amines 2 in fair to excellent yields (Table 2, entries 1, 5, 6 and 8). However, electron-rich aniline derivatives proceeded more sluggishly and led to formation of the desired product with only diminished yields (Table 2, entry 4). In contrast, both electron-deficient and moderately electron-rich heteroaromatic N,O-acetals 1c and 1g–j were readily converted to the corresponding amines 2c, 2g–j (Table 2, entries 3, 7–10), thus giving access to compounds with potential applications in drug design.
|
|
|||
|---|---|---|---|
| Entry | Ar | Yield (%)b | Product |
| a All reactions were performed according to the optimized procedure. b After flash chromatography. c Use of 3 eq. MeMgBr. d Without further purification. | |||
| 1 | 3-ClC6H4 | 87 | 2a |
| 2 | C6H5 | 63 | 2b |
| 3 | 4-pyridyl | 81 | 2c |
| 4 | 4-OMeC6H4 | 40 | 2d |
| 5 | 4-COOEtC6H4 | 94c | 2e |
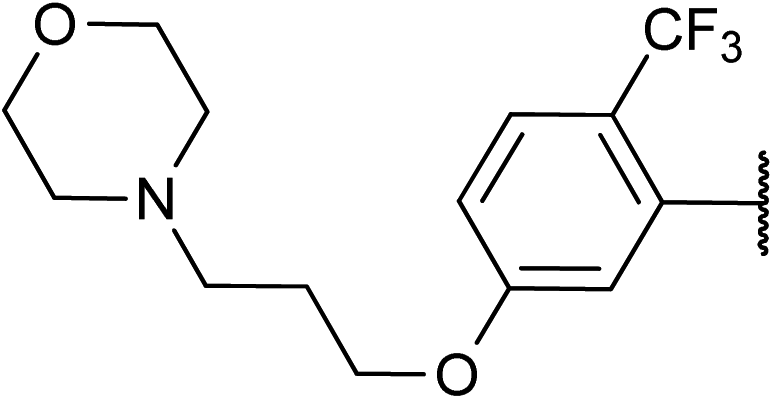
| 6 |
|
57 | 2f |
| 7 |

|
69 | 2g |
| 8 |

|
96d | 2h |
| 9 |

|
80 | 2i |
| 10 |
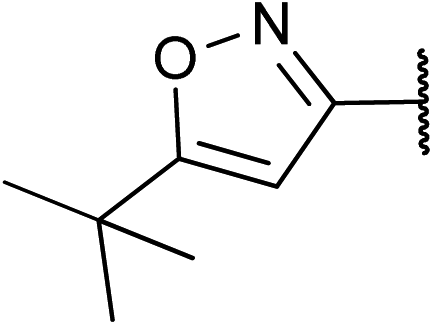
|
74 | 2j |

Next, we turned our attention to other Grignard reagents for nucleophilic addition to trifluoromethylated N,O-acetals. Thus, upon treatment of 3-chlorophenyl hemiaminal 1a with several alkyl Grignard reagents, various α-branched trifluoromethyl N-arylamines 3a–e were obtained in moderate to good yields (Table 3, entries 1–6). Notably, even highly sterically hindered nucleophiles like t-BuMgCl or cyclohexylmagnesium bromide can be successfully employed in this reaction (Table 3, entries 4 and 5). Interestingly, by using i-PrMgCl, i-PrMgCl·LiCl and cyclohexylmethylmagnesium chloride as nucleophiles, also generation of the formal reduction product 3-chloro-N-(2,2,2-trifluoroethyl)aniline 4 was observed. It is worth mentioning that the yield of this side-product was substantially higher with i-PrMgCl·LiCl (up to 30%) than with i-PrMgCl and cyclohexylmethylmagnesium chloride (Table 3, entries 1, 2 and 6). This is presumably due to the higher degree of complexation in the presence of LiCl, which facilitates hydride transfer to the substrate. Furthermore, nucleophilic addition of alkenyl Grignard reagents proceeded smoothly and provided the desired unsaturated trifluoromethyl N-arylamines 3f, 3g and 3h in moderate to good yields (Table 3, entries 7–9).
|
|
|||
|---|---|---|---|
| Entry | RMgX | Yield of 3 (%)b | Yield of 4 (%)b |
| a All reactions were performed according to the optimized procedure. b After flash chromatography. c nd = not detected. | |||
| 1 | i-PrMgCl | 3a, 70 | 16 |
| 2 | i-PrMgCl·LiCl | 3a, 67 | 30 |
| 3 | n-BuMgBr | 3b, 94 | ndc |
| 4 | t-BuMgCl | 3c, 57 | nd |
| 5 |

|
3d, 78 | nd |
| 6 |

|
3e, 26 | 19 |
| 7 |

|
3f, 62 | nd |
| 8 |
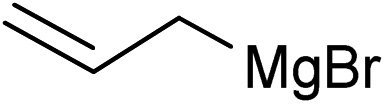
|
3g, 84 | nd |
| 9 |

|
3h, 85 | nd |

Finally, PhMgCl can be used for conversion of trifluoromethyl N,O-acetals into trifluoromethylated benzylamine derivatives. For instance, treatment of the pyrazine derivative 1i and the isoxazolyl hemiaminal ether 1j with 2 eq. PhMgCl afforded amines 5 and 6 in good yields (Scheme 2).
 | ||
| Scheme 2 | ||
In summary, an efficient procedure for the synthesis of α-branched trifluoromethylated amines has been developed starting from stable N-aryl trifluoromethyl hemiaminal ethers. Whereas alkyl amines were incompatible with N,O-acetal formation, a broad range of aromatic and heteroaromatic substrates can be applied successfully to allow for rapid generation of functionalized amine scaffolds for medicinal chemistry purposes after addition of alkyl, alkenyl and aryl Grignard reagents. Moreover and in contrast to other known protocols, protecting group manipulations are not required if the resulting trifluoromethylated amines are to be used as amide bio-isosteres for use in lead optimization. Further investigations in this direction and on the use of functionalized organometallic reagents are ongoing and will be reported in due course.
Acknowledgements
This work was supported by the Excellence Cluster CIPSM and the Deutsche Forschungsgemeinschaft. We thank Mr Christian von der Heyden and Mr Markus Knoth (Merck) for NMR support and fruitful discussions.Notes and references
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- M. Morgenthaler, E. Schweizer, A. Hoffmann-Röder, F. Benini, R. E. Martin, G. Jaeschke, B. Wagner, H. Fischer, S. Bendels, D. Zimmerli, J. Schneider, F. Diederich, M. Kansy and K. Müller, ChemMedChem, 2007, 2, 1100 CrossRef CAS PubMed.
- (a) M. Sani, A. Volonterio and M. Zanda, ChemMedChem, 2007, 2, 1693 CrossRef CAS PubMed; (b) A. Volonterio, S. Bellosta, F. Bravin, M. C. Bellucci, L. Bruché, G. Colombo, L. Malpezzi, S. Mazzini, S. V. Meille, M. Meli, C. Ramírez de Arellano and M. Zanda, Chem. – Eur. J., 2003, 9, 4510 CrossRef CAS PubMed; (c) M. Zanda, New J. Chem., 2004, 28, 1401 RSC; (d) M. Molteni, C. Pesenti, M. Sani, A. Volonterio and M. Zanda, J. Fluorine Chem., 2004, 125, 1735 CrossRef CAS.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed.
- J. Gante, Angew. Chem., Int. Ed., 1994, 33, 1699 CrossRef.
- (a) W. C. Black, C. I. Bayly, D. E. Davis, S. Desmarais, J.-P. Falguyret, S. Léger, F. Massé, D. J. McKay, J. T. Palmer, M. D. Percival, J. Robichaud, N. Tsou and R. Zamboni, Bioorg. Med. Chem. Lett., 2005, 15, 4741 CrossRef CAS PubMed; (b) J. Y. Gauthier, N. Chauret, W. Cromlish, S. Desmarais, L. T. Duong, J.-P. Falgueyret, D. B. Kimmel, S. Lamontagne, S. Léger, T. LeRiche, C. S. Li, F. Massé, D. J. McKay, D. A. Nicoll-Griffith, R. M. Oballa, J. T. Palmer, M. D. Percival, D. Riendeau, J. Robichaud, G. A. Rodan, S. B. Rodan, C. Seto, M. Thérien, V.-L. Truong, M. C. Venuti, G. Wesolowski, R. N. Young, R. Zamboni and W. C. Black, Bioorg. Med. Chem. Lett., 2008, 18, 923 CrossRef CAS PubMed.
- Review about CF3 containing scaffolds: J. Nie, H. C. Guo, D. Cahard and J. A. Ma, Chem. Rev., 2011, 111, 455 CrossRef CAS PubMed.
- For selected examples see: (a) F. Gosselin, P. D. O'Shea, S. Roy, R. A. Reamer, C.-Y. Chen and R. P. Volante, Org. Lett., 2005, 7, 355 CrossRef CAS PubMed; (b) M.-W. Chen, Y. Duan, Q.-A. Chen, D.-S. Wang, C.-B. Yu and Y.-G. Zhou, Org. Lett., 2010, 12, 5075 CrossRef CAS PubMed; (c) A. Henseler, M. Kato, K. Mori and T. Akiyama, Angew. Chem., Int. Ed., 2011, 50, 8180 CrossRef CAS PubMed; (d) Z.-J. Liu and J.-T. Liu, Chem. Commun., 2008, 5233 RSC.
- Y. Gong, K. Kato and H. Kimoto, Bull. Chem. Soc. Jpn., 2002, 75, 2637 CrossRef CAS.
- For selected examples see: (a) M. Liu, J. Li, X. Xiao, Y. Xie and Y. Shi, Chem. Commun., 2013, 49, 1404 RSC; (b) V. A. Soloshonok and T. Ono, J. Org. Chem., 1997, 62, 3030 CrossRef CAS PubMed; (c) Y. Wu and L. Deng, J. Am. Chem. Soc., 2012, 134, 14334 CrossRef CAS PubMed.
- For selected examples see: (a) D. Enders and K. Funabiki, Org. Lett., 2001, 3, 1575 CrossRef CAS PubMed; (b) G. K. S. Prakash, M. Mandal and G. A. Olah, Angew. Chem., 2001, 113, 609 CrossRef; (c) H. Kawai, A. Kusuda, S. Nakamura, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2009, 48, 6324 CrossRef CAS PubMed; (d) V. L. Truong and J. Y. Pfeiffer, Tetrahedron Lett., 2009, 50, 1633 CrossRef CAS; (e) V. L. Truong, M. S. Ménard and I. Dion, Org. Lett., 2007, 9, 683 CrossRef CAS PubMed; (f) P. Fu, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 5530 CrossRef CAS PubMed; (g) F. Gosselin, A. Roy, P. D. O'Shea, C.-y. Chen and R. P. Volante, Org. Lett., 2004, 6, 641 CrossRef CAS PubMed; (h) J. Legros, F. Meyer, M. Coliboeuf, B. Crousse, D. Bonnet-Delpon and J.-P. Bégué, J. Org. Chem., 2003, 68, 6444 CrossRef CAS PubMed.
- For selected examples see: (a) P. Bravo, M. Guidetti, F. Viani, M. Zanda, A. L. Markovsky, A. E. Sorochinsky, I. V. Soloshonok and V. A. Soloshonok, Tetrahedron, 1998, 54, 12789 CrossRef CAS; (b) S. Fries, J. Pytkowicz and T. Brigaud, Tetrahedron Lett., 2005, 46, 4761 CrossRef CAS.
- C. Lauzon and A. B. Charette, Org. Lett., 2006, 8, 2743 CrossRef CAS PubMed.
- (a) F. Grellepois, A. Ben Jamaa and A. Gassama, Eur. J. Org. Chem., 2013, 6694 CrossRef CAS; (b) F. Grellepois, J. Org. Chem., 2013, 78, 1127 CrossRef CAS PubMed Related work with organomagnesium compounds: S. D. Kuduk, C. N. D. Marco, S. M. Pitzenberger and N. Tsou, Tetrahedron Lett., 2006, 47, 2377 Search PubMed.
- A. Ishii, K. Higashiyama and K. Mikami, Synlett, 1997, 1381 CrossRef CAS.
- Related work with (a) organolithium compounds: A. Ishii, F. Miyamoto, K. Higashiyama and K. Mikami, Tetrahedron Lett., 1998, 39, 1199 CrossRef CAS Related work with (b) organozinc compounds: H. Xiao, Y. Huang and F.-L. Qing, Tetrahedron: Asymmetry, 2010, 21, 2949 CrossRef.
- T. Johnson and M. Lautens, Org. Lett., 2013, 15, 4043 CrossRef CAS PubMed.
- Y. Gong and K. Kato, J. Fluorine Chem., 2004, 125, 767 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c3ra47708h |
| This journal is © The Royal Society of Chemistry 2014 |