
Direct conjugation of DNA to quantum dots for scalable assembly of photoactive thin films†
Abstract
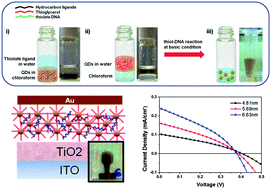
For many thin film applications, it is critical to not only control the organization of the materials on surfaces but to also use scalable processes that are time and material efficient. This is especially the case when using nanomaterials, including metal or semiconductor nanocrystals, as building blocks from which to engineer devices. In this work, we demonstrate a method to directly conjugate DNA to cadmium based quantum dots (QD) to create thin film arrays on surfaces for potential optoelectronic devices. These DNA-conjugated QDs showed uniform coatings, were oxidation-stable, and remained stable in high ionic strength environments. In previously published work, we discovered that high salt, in particular magnesium, is critical for fabricating nanoparticle assemblies on substrates through DNA interactions. The QD thin films were produced by means of interparticle DNA hybridization in a few steps with no loss of material and with good control over film thickness and roughness. By directly conjugating the DNA to the QDs, it also became possible to study DNA's role in mediating charge transport in the QD films. For this, DNA-conjugated CdTe nanocrystals were assembled onto TiO2 films to fabricate photovoltaic prototypes. Current–Voltage measurements from the DNA–QD devices showed the promise of using DNA not only as an assembler but also as mediator of charge separation and transport.
 Please wait while we load your content...
Please wait while we load your content...

