 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
XPS and NEXAFS study of fluorine modified TiO2 nano-ovoids reveals dependence of Ti3+ surface population on the modifying agent†
Jan-Yves Ruzickaa,
Faridah Abu Bakarab,
Lars Thomsenc,
Bruce C. Cowiec,
Campbell McNicollade,
Tim Kemmittde,
Helen E. A. Brandc,
Bridget Inghamde,
Gunther G. Anderssonf and
Vladimir B. Golovko*ae
aUniversity of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand. E-mail: vladimir.golovko@canterbury.ac.nz
bUniversiti Tun Hussein Onn Malaysia, 86400 Parit Raja, Batu Pahat, Johor, Malaysia
cAustralian Synchrotron, 800 Blackburn Road, Clayton, Melbourne, Australia
dCallaghan Innovation, PO Box 31310, Lower Hutt 5040, New Zealand
eThe MacDiarmid Institute for Advanced Materials and Nanotechnology, PO Box 600, Wellington 6140, New Zealand
fFlinders Centre for NanoScale Science and Technology, Flinders University, PO Box 2100, Adelaide SA 5001, Australia
First published on 15th April 2014
Abstract
Crystalline titanium dioxide was synthesised under mild conditions by the thermal degradation of peroxotitanic acid in the presence of a number of fluoride-containing surface modifying agents (NH4F, NH4BF4, NH4PF6, NBu4F, NBu4BF4, NBu4PF6). The resulting materials were characterised by PXRD, SEM, HRTEM, XPS and NEXAFS. Particle phase, size, and surface area were noticeably affected by the choice of surface modifying agent. Both the cation and anion comprising the modifying agent affect the surface Ti3+ population of the materials, with two apparent trends observed: F− > BF4− > PF6− and NBu4+ > NH4+. All materials displayed evidence of fluorine doping on their surfaces, although no evidence of bulk doping was observed.
1 Introduction
Titanium dioxide (titania) is one of the most well-studied of the known photocatalysts1 due to its wide band gap, chemical and biological inertness, non-toxicity, and commercial availability.2 Since Fujishima and Honda's work demonstrating its photocatalytic activity,3 many researchers have focussed on developing the catalyst (particularly the anatase phase) for applications in a wide variety of fields, including photocatalytic air and water remediation,4–6 self-cleaning surfaces,4 photovolatics,2,5,6 water splitting,5,6 and sensors.5 Nanosized titanium dioxide is of particular interest to researchers, due to its high surface area compared to macroscale catalyst. This allows for the exposure of orders of magnitude more active sites than would be available on bulk material.5However, titanium dioxide exhibits three features that limit its usefulness as a photocatalyst:
(1) Its wide band gap limits light absorption to photons with energy >3.2 eV (for anatase),7 which represents a very small fraction of available solar light.8
(2) Photocatalytic efficiency is limited by the recombination of exciton pairs,2 especially for doped or poorly-crystalline samples.
(3) Titanium dioxide nanocrystals thermodynamically favour low-energy crystal faces (the {101} face for anatase, for example), which are less catalytically active than higher-energy faces.9
Much recent research has focussed on the mitigation of these drawbacks. Anion doping has recently seen use as a means of narrowing the anatase band gap by introduction of mid-gap states; the popularity of this technique should be attributed to Asahi et al. for their work on nitrogen-doping of titania.10 Recombination events can be limited either by increasing the crystallinity of the catalyst (for example, through high-temperature treatment)11 or by encouraging charge separation.12 The industrial catalyst Aeroxide P-25, for example, may discourage recombination events by the inclusion of rutile-phase titania, which acts as an electron trap and efficiently separates charge.12
The problem of facial selectivity in titanium dioxide nanocrystal growth (particularly in the case of the sol–gel synthetic pathway) was recently addressed by Lu et al., who showed that the fluoride ion would preferentially bind to the high-energy anatase {001} face.9 In this way the fluoride acted as a surface modifying agent (SMA) and encouraged the growth of this crystal face even under mild conditions. Surface-bound fluoride has also been shown to decrease charge recombination by acting as an electron trap,13 and may even narrow the titania band gap when used as a dopant.14 Thus, titanium dioxide synthesis in the presence of fluoride shows excellent promise as a means of addressing the problems currently faced in sol–gel titanium dioxide synthesis.
While the breakthrough paper by Lu et al. paved the way for a number of studies on the synthesis of high-energy titania nanocrystals,15–22 their original work (and many follow-up papers) focussed on the use of toxic and highly dangerous hydrofluoric acid as a fluoride source, combined with high-pressure/high-temperature synthetic conditions. Some researchers (notably Yu et al.23) have shown similar results using ionic liquids, which are considerably more complex than the relatively simple fluoride sources used previously. Only very recently have researchers focussed on simpler and less toxic sources of fluoride or fluoride-containing species.24–33 To the authors' knowledge, there currently exists no systematic study of such fluoride sources and their effects on the synthesis of titanium dioxide nanoparticles.
Here we present a study of the effect of a number of fluoride-containing salts (NH4F, NH4BF4, NH4PF6, NBu4F, NBu4BF4, NBu4PF6) on the synthesis of crystalline titanium dioxide nanoparticles via the thermal degradation of peroxotitanic acid under mild conditions. We have purposefully selected a wide range of fluoride-containing surface modifying agents to gauge the effect not only of the fluoride source (F−, BF4−, PF6−) but also of the cation (NH4+, NBu4+) on the formation of titanium dioxide nanoparticles. While some of these salts (i.e. NH4F,25,27,34 NH4BF4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26) have seen previous use in literature, the majority of the modifying agents used in this study have not been tested previously. We report the effect of SMA on particle size, crystallinity and surface area, as well as Ti3+ population.
26) have seen previous use in literature, the majority of the modifying agents used in this study have not been tested previously. We report the effect of SMA on particle size, crystallinity and surface area, as well as Ti3+ population.
2 Results and discussion
2.1 Particle sizing
As-prepared materials were characterised by scanning electron microscopy (SEM) and dynamic light scattering (DLS) to determine particle size. A minimum of 200 particles were analysed for each sample for SEM particle size counting. Particle sizes are shown in Fig. 1, sample SEM images of particles are shown in Fig. 2 (for comparison SEM image of titania synthesised in the absence of SMA, see Fig. S1†).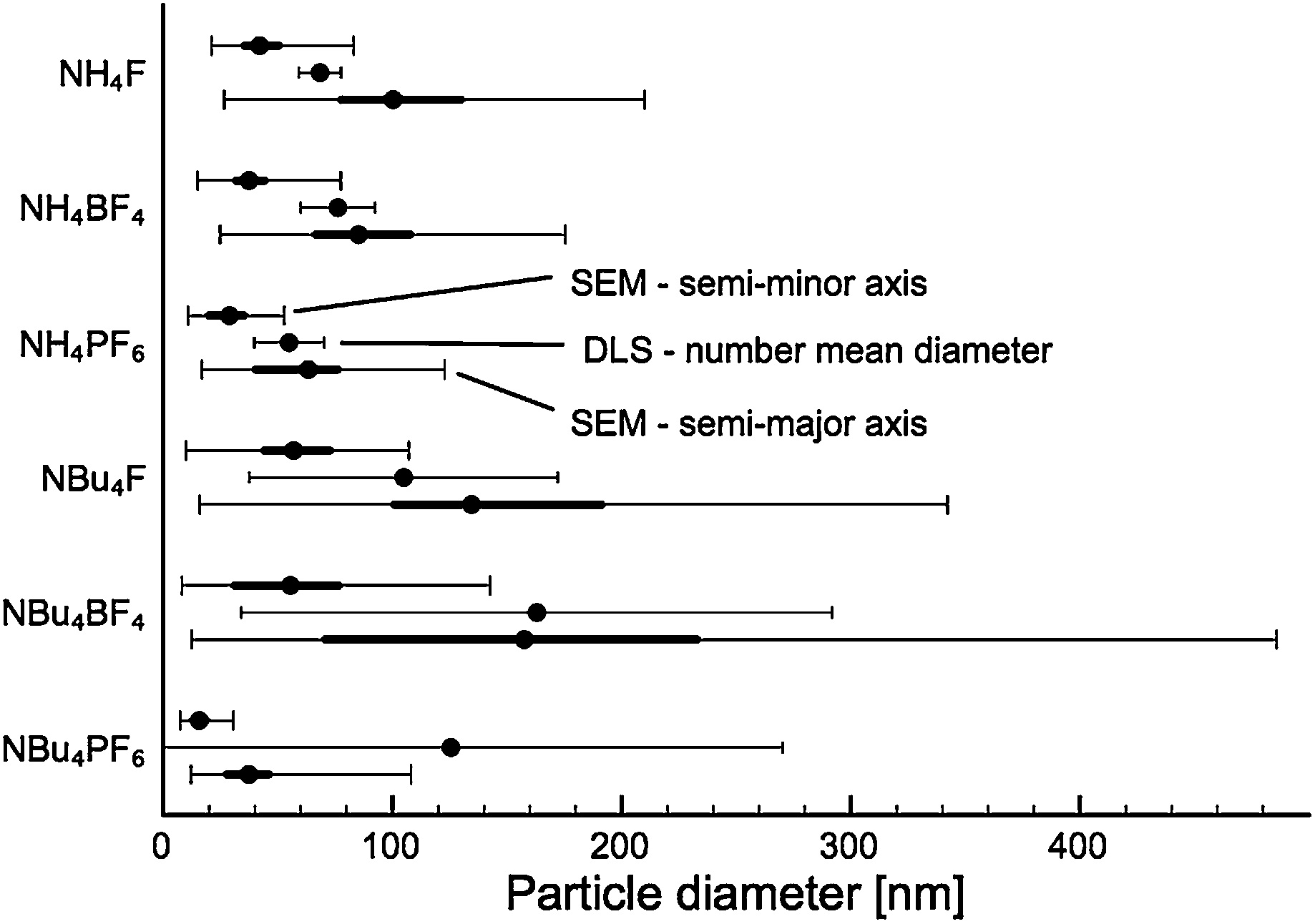 | ||
| Fig. 1 Particle diameter measured along the semi-major and semi-minor axes, as well as average particle diameter measured by DLS. For SEM particle sizing, the thin line indicates the full range of particles counted, the thick line the upper and lower quartiles, and the central dot the median value. For DLS the dot indicates the number-mean particle size and the error bars the 95% confidence interval. | ||
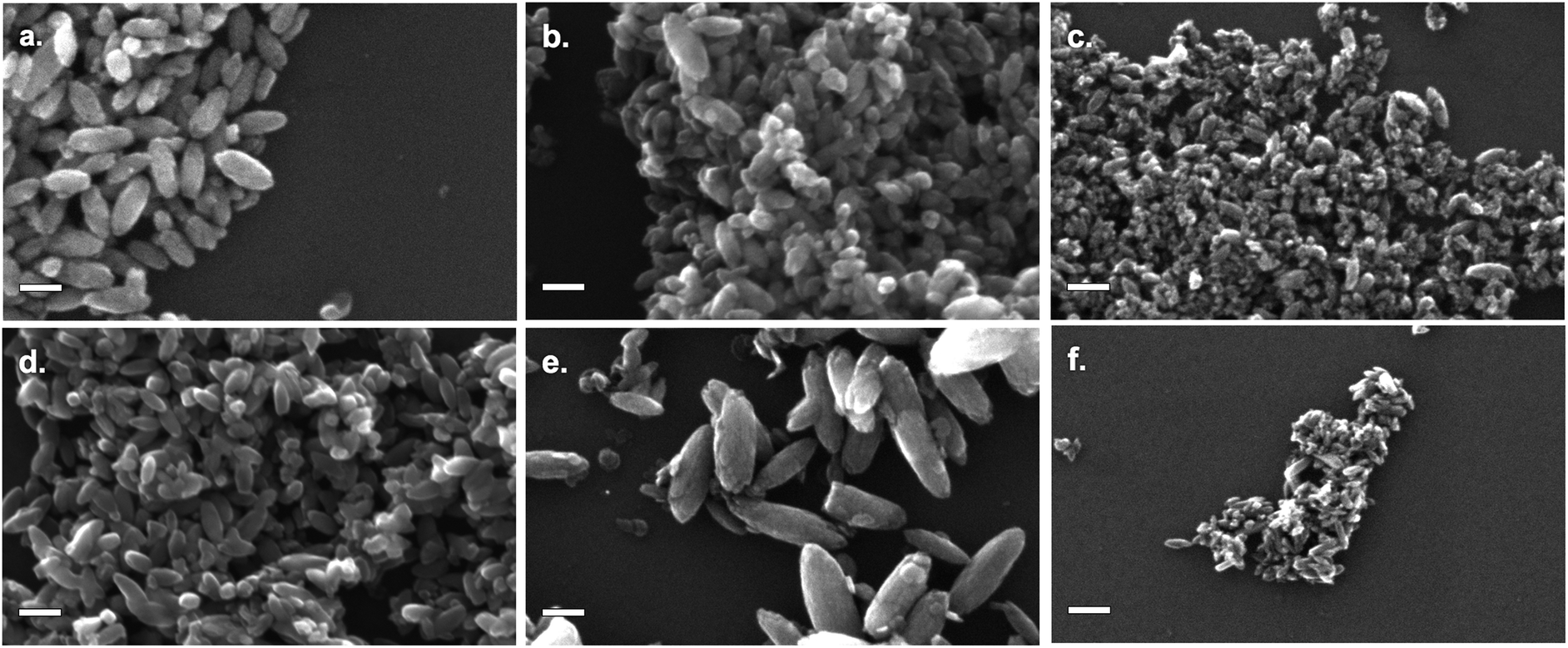 | ||
| Fig. 2 SEM images of TiO2 modified by: (a) NH4F; (b) NH4BF4; (c) NH4PF6; (d) NBu4F; (e) NBu4BF4; (f) NBu4PF6. All scale bars are 100 nm. | ||
All products were found to be ovoid nanoparticles, with a mean anisotropy (calculated by SEM particle sizing) of 0.40–0.49. DLS data generally agreed with SEM particle sizes, except for NBu4PF6-modified TiO2. The presence of NBu4PF6 on the surface of the particles likely encourages their aggregation in solution, giving rise to a much larger apparent size by DLS.
While particles synthesised in the presence of F− and BF4− appear to have approximately the same size, particles synthesised in the presence of PF6− are much smaller. Of note, there appears to be a considerable difference in size between NH4BF4− and NBu4BF4-modified TiO2. Specifically, particles synthesised in the presence of NBu4-containing SMAs are much more polydisperse. This indicates that particle size is dependent not only on the anionic group, but also on the cationic group of the SMA.
2.2 Surface area measurements
Surface area measurements (BET isotherm) are shown in Fig. 3. All samples exhibited type 4 isotherms, with some small hysteresis suggesting larger pores (adsorption curves shown in Fig. S2†). | ||
| Fig. 3 BET surface areas for TiO2 modified by different fluoride-containing surface modifying agents. | ||
2.3 Crystallinity
High-quality PXRD patterns were collected at the powder diffraction beamline at the Australian Synchrotron. Full-pattern fitting analysis of PXRD data showed that all products were crystalline, and generally formed the anatase phase of titania (Fig. 4): in fact, materials synthesised in the presence of NH4+-containing surface modifying agents showed no rutile or brookite content. Titanium dioxide synthesised by a similar manner in the absence of fluoride ions (Fig. S3†) forms a roughly 28:72 mixture of anatase and rutile. Yu and coworkers35 have previously observed this effect, and argued that the presence of fluoride may either prevent phases other than anatase from forming (due to preferential binding) or may encourage the phase transformation to anatase. For NBu4+–SMA-modified materials, rutile and even brookite were observed (see Table 1 for exact amounts). If it is assumed that phase selectivity is due to the presence of SMA, the presence of these phases suggests that in the case of NBu4+–SMAs, the anion is not as influential as the cation in the formation of the nanoparticle.
 | ||
| Fig. 4 PXRD diffraction patterns for TiO2 modified with fluoride salts. Anatase, rutile and brookite theoretical peak positions are shown at the bottom of the graph. The abscissa is given in momentum transfer units Q = 4πsin(θ)/λ. | ||
| SMA | Anatase | Rutile | Brookite | |||
|---|---|---|---|---|---|---|
| Fraction [%] | Dimensions [nm] | Fraction [%] | Dimensions [nm] | Fraction [%] | Dimensions [nm] | |
| a Sample is isotropic in shape. | ||||||
| NH4F | 100 | 28.7 ± 1.3 × 119 ± 12 | ||||
| NH4BF4 | 100 | 27.2 ± 1.2 × 90 ± 9 | ||||
| NH4PF46 | 49 ± 4 | 19.8 ± 0.7a | ||||
| 51 ± 4 | 3.3 ± 0.2a | |||||
| NBu4F | 76 ± 3 | 70 ± 8 × 200 ± 30 | 24 ± 3 | 13 ± 2a | ||
| NB4BF4 | 48.5 ± 1.6 | 11.8 ± 0.7 × 40 ± 3 | 43.6 ± 1.0 | 16.8 ± 0.6 × 38.3 ± 1.4 | 7.9 ± 0.9 | 8.8 ± 1.2a |
| NBu4PF6 | 57.2 ± 1.4 | 10.9 ± 0.5 × 34.1 ± 1.7 | 22.4 ± 1.1 | 8.2 ± 0.8 × 20.9 ± 0.8 | 20.5 ± 0.9 | 6.8 ± 0.4a |
The titanium occupancy was allowed to vary during the Rietveld refinement. In most cases the titanium occupancy of the materials was less than one, i.e. the crystal lattice was titanium deficient. Titanium-deficient titanium dioxide has previously been attributed to the presence of OH− ions within the bulk.37 Given the use of H2O2 in the synthesis of the products reported here, it is feasible that OH− groups would be trapped within the lattice, resulting in a lower-than-expected titanium occupancy. The titanium occupancy within the anatase phase was approximately 0.82 for all NH4+–SMA-modified products and 0.89 for NBu4+–TiO2. The small difference in Ti deficiencies between the two cation types of SMAs suggests the Ti deficiency is only partially dependent on the SMA but mostly due to the peroxo-titanic acid hydrothermal synthesis. Rutile in NBu4BF4–TiO2 and NBu4PF6–TiO2 exhibited titanium occupancy of 0.88: very similar to the majority of anatase phase crystallites, suggesting again that deficiencies in Ti are related to synthesis conditions rather than the SMA.
Size and microstrain analysis of the PXRD data by Rietveld refinement (Fig. S4†) shows that a majority of the products are anisotropic in shape. All samples except NH4PF6–TiO2 show preferential orientation for both anatase and rutile in the [001] direction, and are also anisotropically elongated in this direction. Additionally, the anatase (004) peak is both larger and narrower than expected from shape- and direction-isotropic modelling of a population of the same crystallite size. These peak profiles are best explained by rod-like crystallites oriented along the [001] direction. The larger area is explained by the preferred orientation, and the sharper line profile is explained by increased crystallinity along the major planes of the rod (see Fig. S5†). As can be seen in Table 1 the addition of SMA produced anatase crystallites that were elongated in the [001] direction for all cases except NH4PF6–TiO2. The dimensions observed in SEM and those calculated from PXRD data largely agree. The exception is NBu4BF4–TiO2, whose ovoid particles (observed in SEM) have dimensions far larger than those determined by PXRD. It is likely that the observed particles consist of multiple grains.
For NH4PF4–TiO2, the PXRD data was best modelled by two isotropic (in shape and direction) anatase populations, one population of diameter ∼3.3 nm and another larger population of diameter 19.8 nm. The larger anatase population showed no preferred crystallite orientation, although in SEM the larger particles appeared elongated (Fig. 2C). The aggregation of smaller particles around the larger particles could have prevented them from preferentially orienting, or alternatively the observed particles may not be single crystallites. The rutile phase titania also showed rod-like character and exhibited preferential orientation (however, for NBu4F–TiO2 particle shape could not be determined due to the lack of definition in rutile peaks). This rod-like form is typical of rutile grown in an acidic environment.38,39
HRTEM studies further confirmed the particle size and shape model developed based on SEM and PXRD data, demonstrating strong fringing patterns (Fig. 5) indicative of crystalline material. Ovoid particles exhibited strong fringing indicative of the [101] plane, at approximately 45° to their long edge, suggesting that their shape was dictated by the growth of specific crystal faces. Based on this data, it is proposed that the (010) face is located on the long edge of the nanoparticle (Fig. 6).40 It should be noted that many smaller particles do not follow this pattern: these particles appear more regularly in products synthesised using NBu4-containing SMAs.
 | ||
| Fig. 5 HRTEM fringing from the (101) face of fluorine-modified TiO2 proceeds at ∼45° to the long edge of the nanoparticle. (Shown here: TEM image of NH4F–TiO2.) | ||
 | ||
| Fig. 6 The proposed structure of nanocrystalline TiO2. (Shown here: NBu4F–TiO2.) | ||
2.4 Electron spectroscopy studies
X-ray photoelectron spectroscopy (XPS) was performed at the soft X-ray beamline of the Australian Synchrotron.41 Survey scans (see ESI, Fig. S6†) showed evidence of silicon (from the wafer), carbon, titanium, oxygen, nitrogen, and fluorine, with boron and phosphorus observed for BF4−- and PF6−-modified materials (ESI, Fig. S7†). High-resolution XPS was performed on the fluorine 1s, titanium 2p, nitrogen 1s, oxygen 1s and carbon 1s peaks, as well as the phosphorus 2p peak for PF6−-modified samples. Peak positions, intensities, and FWHMs are tabulated in ESI, Tables S1 and S2.†Two oxygen 1s peaks were observed (see Fig. 7 for a typical O 1s XP spectrum). The peak at ∼532.5 eV was assigned to surface Ti–O–H and SiO2, while the one at ∼530 eV were assigned to Ti–O–Ti.42 In all cases the Ti:O ratio by XPS was approximately 1:2, suggesting that any deviation in occupancy levels in the bulk did not continue to the surface of the material.
 | ||
| Fig. 7 A typical O 1s XP spectrum (shown here: NH4F–TiO2), with peak assignments. | ||
The presence of fluoride was confirmed by XPS (Fig. 8, fluorine populations for all samples shown in ESI, Fig. S8†). Two peaks were observed: a broad peak at 687–688 eV (A) and a sharper one at 684–685 eV (B). All samples exhibited this double-peak feature, despite being washed several times with distilled water (and additionally with acetonitrile for tetrabutylammonium salt-modified samples), indicating that the SMA was strongly bound to the surface. While fluorine peaks in the region of peak A have previously been attributed to surface lattice fluoride (Ti–F–Ti),13,26,43–45 it was found that this peak could be removed by treatment with KOH solution (1 M, one hour: Fig. 8). This suggests that the fluoride species is more likely to be surface-bound, as lattice fluorine has previously been reported to be stable to base treatment.43 Additionally, a small carbon XPS peak (see ESI, Fig. S9†) is observed to disappear after KOH treatment: this peak may be attributable to fluorocarbon species.46 Peak B is assigned to surface-bound, chemisorbed Ti–F groups based on its binding energy position.13,43,44 However, KOH treatment does not remove the species giving rise to peak B, although washing with less concentrated base solutions16,47 has previously been shown to remove chemisorbed Ti–F groups. The reason for the stability of this species even under basic conditions is as yet unknown.
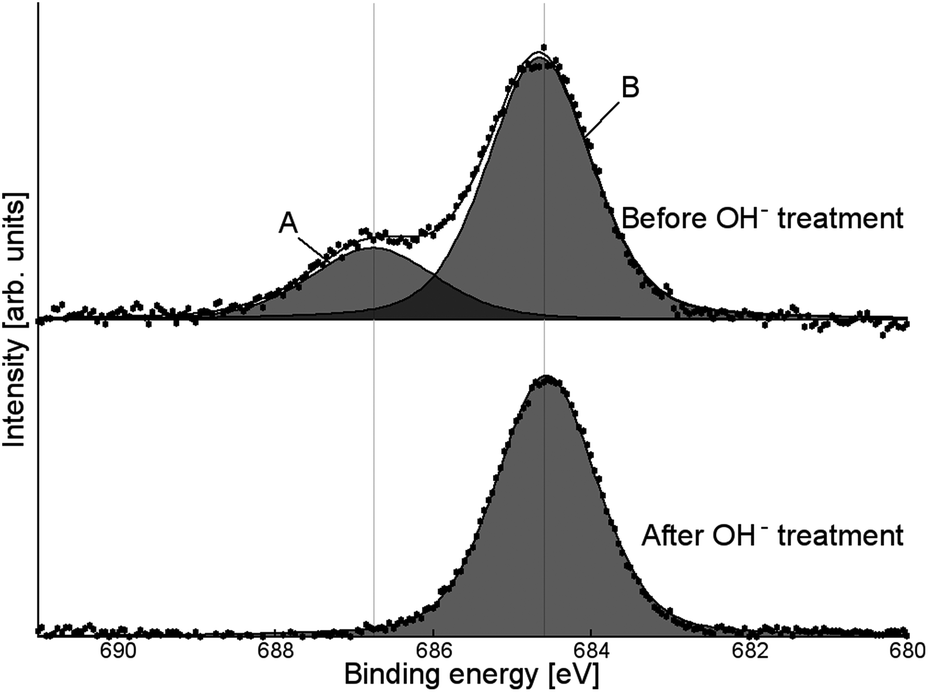 | ||
| Fig. 8 High-resolution F XPS signal for NH4PF6-modified TiO2, before and after washing with KOH. | ||
Analysis of the titanium XPS signal (see ESI, Fig. S10† for full fitting) shows a characteristic peak at 459 eV attributed to Ti4+, with a smaller peak around 457.7–458 eV indicative of surface Ti3+. The Ti3+ peak is visible even for untreated P-25 (where it is approximately 1% of the total Ti signal), which has been found before on unmodified TiO2 nanoparticles,48 anatase nanotubes,49 and doped nanotubes.50 On titania single crystals, Ti3+ has been found after ion bombardment.51,52 The Ti3+ sites at the surface of the nanoparticles, nanotubes and doped nanotubes are stable in air.48–50 For those created by ion bombardment on single crystal surfaces the ref. 51 and 52 do not report whether or not the Ti3+ states induced by ion bombardment are stable in air. In all cases, the Ti3+ surface content for SMA-modified TiO2 (as a function of total Ti content, see Fig. 9) was greater than for P-25. This is likely due to the effect of the surface-bound fluoride, which has been previously shown to encourage Ti3+ formation.42
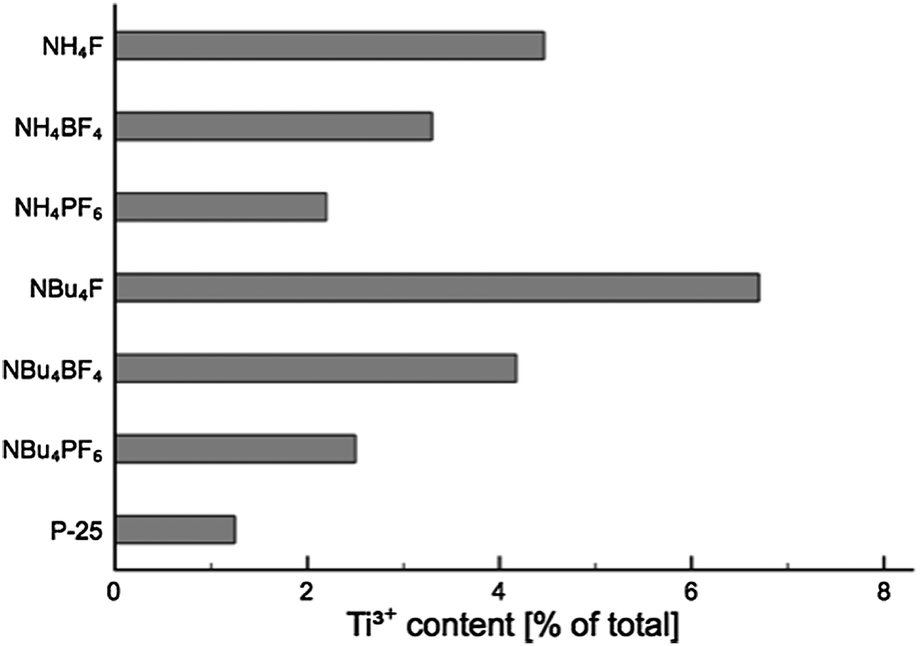 | ||
| Fig. 9 Ti3+ content (as a percentage of total Ti content) for each product. | ||
NBu4F–TiO2 shows the greatest Ti3+ surface concentration, with the Ti3+ XPS signal comprising 7% of the total Ti XPS signal. The exact concentration in any of the layers cannot be determined from a single measurement, however, it is possible to estimate the upper limit for the Ti3+ concentration. Assuming an electron mean free path of 0.38 nm (based on the calculations of Fuentes et al.,53 given Ekin = 360 eV as used for the XPS measurements), 16% of total Ti population of the surface is in the +3 oxidation state in case that Ti3+ is only present in the first monolayer. In this calculation one monolayer is considered as a layer with the thickness of the a-axis of the unit cell of anatase (0.38 nm). Assuming that Ti3+ is distributed equally over the first two or three monolayers, the Ti3+ concentration is calculated be 10% and 9%, respectively. The estimates provided above are solely for the purpose of gauging the upper limit of the Ti3+ concentration at the material surface, and are not meant as a model for the distribution of Ti3+. For determining the distribution of Ti3+ more accurately, XPS measurements over a large range of excitation energies would have to be acquired.
While such a population is considerably higher than that of unmodified TiO2, it is still much lower than could be achieved by, for example, Ne+ ion bombardment, which has been shown to achieve a Ti3+ surface concentration of up to 45%.52
In addition, there is an apparent correlation between both anionic and cationic group of the SMA, and the abundance of Ti3+ in the material (i.e. F− > BF4− > PF6−; NBu4+ > NH4+). The presence of these Ti3+ centres may improve visible light activity through the creation of localised mid-gap states,54,55 aiding in the visible-light activity of the materials.
NEXAFS analysis was performed on the Ti L2,3 transition using Auger and Total Electron Yield (AEY and TEY) and Fluorescence Yield (FY) measurements. Typical AEY, TEY and FY NEXAFS spectra are shown in Fig. 10, and full graphs are available in ESI (Fig. S11†). The AEY spectrum was found to be the most well-defined, with TEY and FY exhibiting less signal. In a majority of cases the edge displays a characteristic shape, with auxiliary peaks marked A and B and main peaks C–G. Peak assignments based on literature reports are shown in Table 2.56,57
 | ||
| Fig. 10 Typical Ti L2,3 NEXAFS spectrum (here: NH4F–TiO2). Peaks referred to in the main text are labelled, and AEY, TEY and FY curves are noted. | ||
| Peak | Assignment |
|---|---|
| A | Satellite peak |
| B | Satellite peak |
| C | t2g L3 |
| D | eg L3 |
| E | eg L3 |
| F | t2g L2 |
| G | eg L2 |
The properties of these peaks (e.g. relative intensities, splitting) may be altered by the presence of dopants in the bulk titania crystal matrix.57 Two particular trends are of interest here:
• The ratio between the peaks C and D is strongly dependent on the crystal phase of the titanium dioxide material, due to long-range structural effects.58 Anatase materials exhibit a strong C and somewhat weaker D peak, while rutile displays the converse.
• The presence of pentacoordinate Ti3+ sites within the bulk of the material results in a greatly decreased intensity in the relative intensities IC/ID and IF/IG, as well as the appearance of a shoulder peak on the low-energy side of peak F.59,60
It can be seen that for NH4+–SMA-modified TiO2 the low-energy peak C is significantly more intense, and the high-energy peak D is apparent only as a shoulder for all scans. For NBu4F- and NBu4PF6-modified TiO2, the high-energy peak is more apparent: this is a result of the presence of significant quantities of rutile in the material, which correlates with the PXRD findings above.
It is also interesting to compare the above-mentioned ratios measured using electron and fluorescence yield. Electron yield NEXAFS is generally sensitive to only the top 50 Ångstroms of the material, while fluorescence yield measurements may penetrate as far as 500–1000 Ångstroms into the bulk.61,62 Thus, comparison of data collected using these techniques allows for a more comprehensive view of the change in particle properties with depth. Graphs of the intensity ratios IC/ID and IF/IG are shown in Fig. 11, and it can immediately be seen that both these ratios increase slightly under FY as compared to AEY and TEY. This suggests that Ti3+ centres are concentrated at the surface of the material, and are considerably less prevalent in the bulk. The only deviations from this pattern are observed for IF/IG in NBu4F and IC/ID for NBu4PF6, in which the FY signals are rather weak.
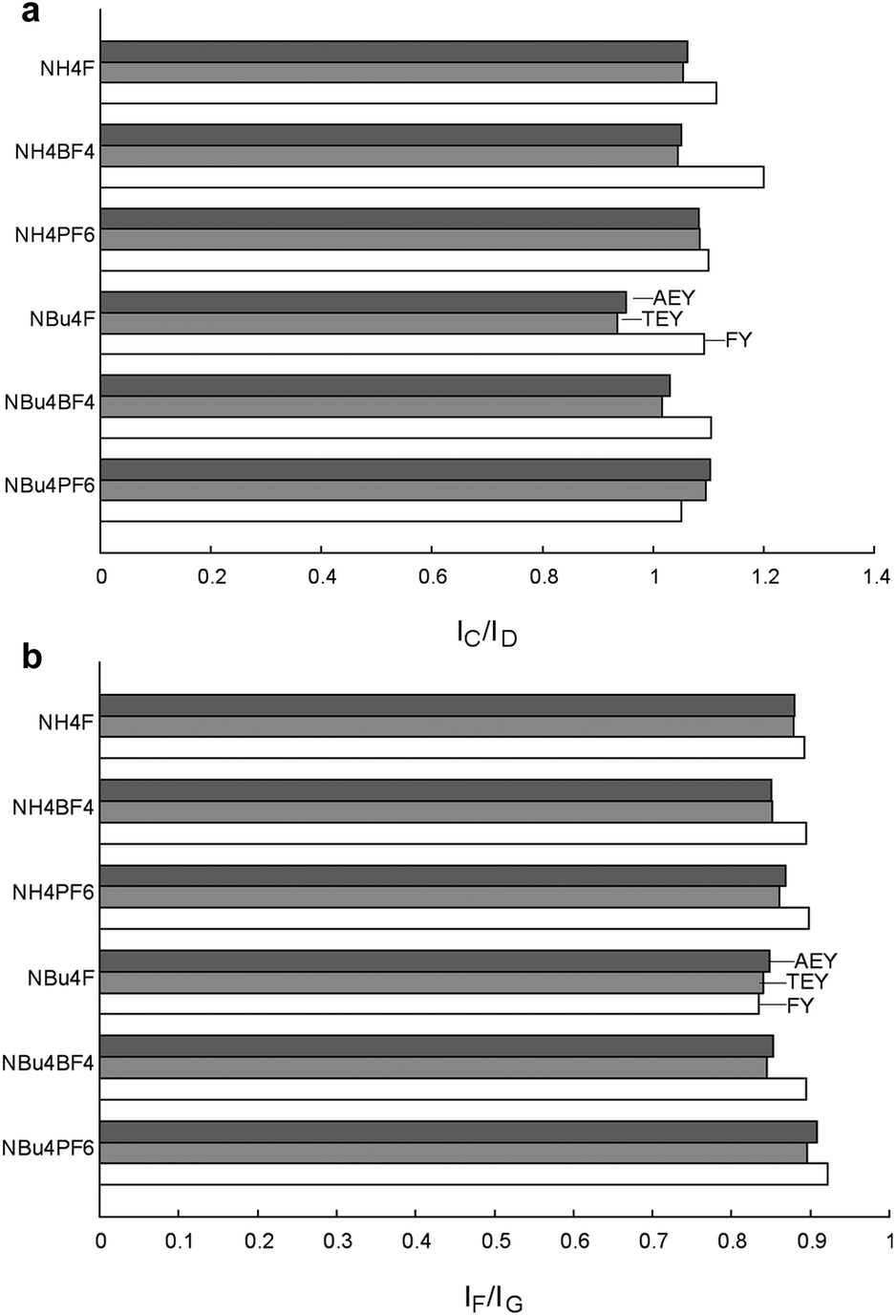 | ||
| Fig. 11 Ratios of NEXAFS peak intensities for fluorine-modified TiO2 materials. Anomalous measurements for NBu4F–TiO2 are due to poor FY data, see ESI Fig. S11† for individual spectra. | ||
2.5 Surface charge
Zeta potential measurements were performed on all samples, without KOH treatment as discussed above (ESI, Fig. S12†). The majority of fluoride-modified materials show a negative zeta potential, the two exceptions being NBu4BF4- and NBu4PF6-modified TiO2. In contrast, TiO2 synthesised in the absence of SMA shows a positive zeta potential. This change in zeta potential is attributed to the fluorinated surface of the modified materials, while the variation in zeta potential suggests that not all SMAs are equal in their ability to influence the surface of the material.There is no correlation between XPS measurements of fluoride intensity (normalised by total Ti signal) and zeta potential. This may be due to the differing preparation methods and media for the two techniques (i.e. measurement in solution at atmospheric pressure as opposed to measurement of the solid under UHV conditions).
2.6 Mechanism of growth
The above results demonstrate that the choice of SMA heavily influences particle size, crystallinity, surface area and electronic structure of the product. In solution, fluoride (F−) will bind strongly to titanium, forming Ti–F. The presence of fluoride bound to titanium during synthesis encourages the growth of anatase (as opposed to rutile) titania:35 this suggests that those materials that exhibit rutile and brookite character are exposed to lower concentrations of fluoride during synthesis. Pure anatase materials exhibited a much larger (004) PXRD peak than predicted by the spherical particle model, which correlated with SEM and TEM observations of anisotropy. This further suggests that the SMA is responsible for alterations in both particle size and particle phase.XPS studies show that fluoride is chemically bound to the TiO2 surface as Ti–F, not as Ti–BF4 or Ti–PF6. Previous reports are inconclusive on the nature of BF4−–SMA modification: Zhou and coworkers recently reported that TiO2 synthesised in acidic NaBF4 solution was modified by the BF4− ion, citing evidence of Ti–B–F species in B XPS;28 other groups have reported that NaBF4 dissociates to give surface Ti–F as an SMA.26,63 It is posited that in the synthetic method outlined in this report, BF4− decomposes to give free fluoride in addition to boric acid (here “![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ti” represents surface-bound titanium):
Ti” represents surface-bound titanium):
| Ti–OH + BF4− → Ti–F + BF3 + OH− |
| BF3 + 3H2O → 3H+ + 3F− + H3BO3 |
PF6− has been reported to decay in a similar manner to give F−.64 We therefore propose a similar mechanism for PF6− in solution:
| Ti–OH + PF6− → Ti–F + PF5 + OH− |
| PF5 + 4H2O → 5H+ + 5F− + H3PO4 |
This mechanism is schematically illustrated in Fig. 12. An increase in temperature accelerates the decomposition of BF4−, especially near the boiling point of the solvent.65 Previously titanium dioxide was synthesised in the presence of BF4− at 130 °C, allowing for the facile production of F−.26 It appears that in the synthetic method reported here, the release of F− is kinetically controlled by the lower temperature of the reaction. If the rate of fluoride formation is significantly slower than the rate of titanium dioxide condensation, the early-stage formation of titanium dioxide will take place in an effectively “fluoride-free” environment.
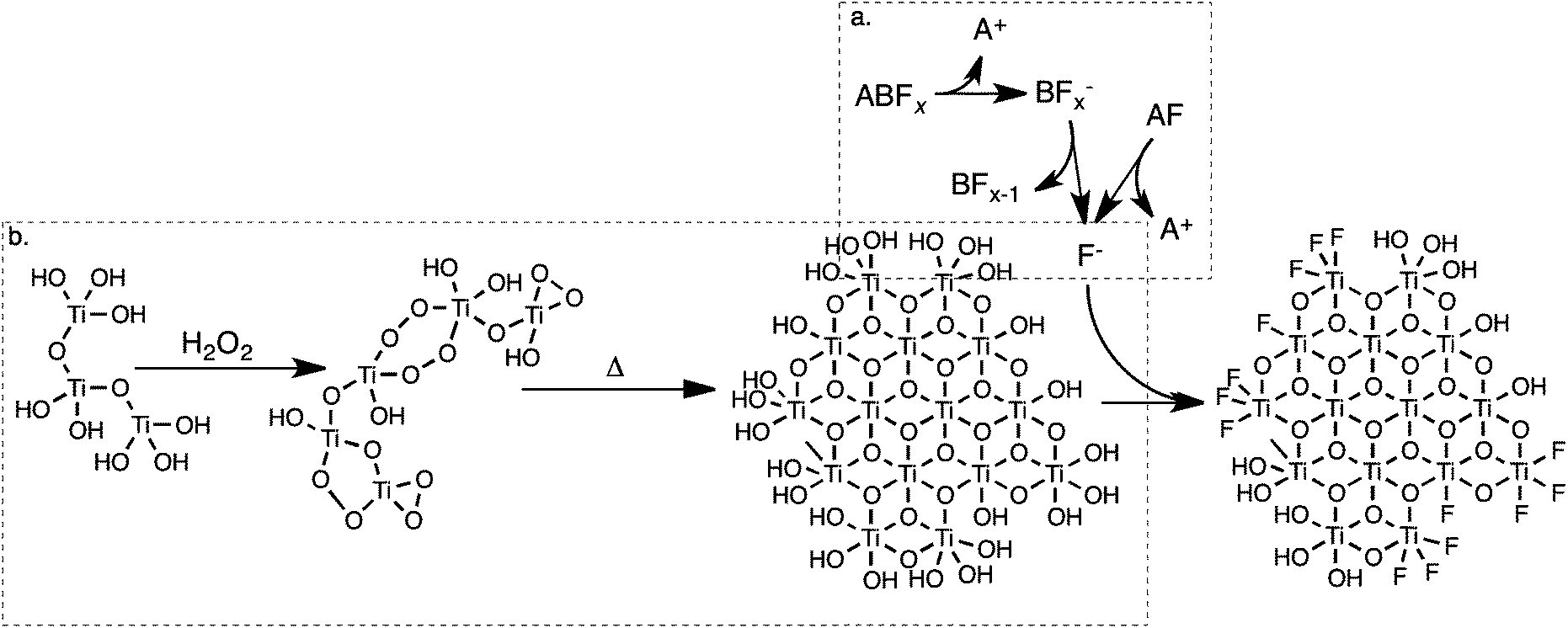 | ||
| Fig. 12 A proposed mechanism for the formation of fluoride-modified TiO2 in solution. F− is formed by the dissolution (and hydrolysis) of fluoride salts (a), while nuclei of titanium dioxide simultaneously form by the breakdown of peroxotitanic acid (b). F− will then chemisorb to the surface of titanium dioxide, dictating its growth. | ||
The presence of rutile and brookite in some materials suggests that during the initial stage of titania formation there is insufficient free fluoride present in solution to ensure complete anatase formation. Non-anatase content appears to increase as anion and cation mass increase. This suggests that PF6− decays at a slower rate than BF4−, and that the presence of NBu4+ as a cation similarly retards the rate of decay.
3 Experimental
Titanium isopropoxide (≥98%, Acros Organics), tetrabutylammonium fluoride (1 M in THF, Acros), ammonium tetrafluoroborate (≥97%, Sigma-Aldrich), ammonium hexafluorophosphate (≥95%, Sigma), tetrabutylammonium hexafluorophosphate (≥98%, Sigma), ammonium fluoride (≥98%, BDH), and tetrabutylammonium tetand tetrabutylammonium rafluoroborate (≥99%, Strem) were all used as supplied, without further purification. P25–TiO2 “Aeroxide” was manufactured by Evonik Degussa GmbH. Titania particles are 80:20 anatase to rutile phase, with a quoted average particle size of 30 nm and surface area of 50 m2 g−1.
3.1 Synthesis
Titanium dioxide was synthesised by thermal degradation of peroxotitanic acid.66 Titanium isopropoxide precursor (1.78 g, 6.27 mmol) was dissolved in ethanol (5 mL) in a 50 mL round-bottom flask with stirring. Milli-Q water (25 mL) was rapidly added with vigorous stirring to the resulting solution at room temperature. This resulted in the rapid hydrolysis of titanium isopropoxide to produce hydrated titanium dioxide, in the form of an opaque white precipitate. The mixture was magnetically stirred (600 rpm) for a further ten minutes to ensure complete hydrolysis, and the precipitate was then collected by centrifugation (5 min at 5000 rpm) and washed several times with Milli-Q water (by repeated redispersion using Vortex agitator followed by centrifugation as before) to remove ethanol and isopropanol formed during hydrolysis. The titanium dioxide powder was redispersed in water (50 mL) and combined with fluoride salt. The following salts were used: ammonium fluoride (NH4F), ammonium tetrafluoroborate (NH4BF4), ammonium hexafluorophosphate (NH4PF6), tetrabutylammonium fluoride (NBu4F), tetrabutylammonium tetrafluoroborate (NBu4BF4), and tetrabutylammonium hexafluorophosphate (NB4PF6). Salts were added in a 1:1 molar ratio with titanium (assuming complete hydrolysis and retention of the solid material during washing). Hydrogen peroxide (4.26 g 50% w/w, 62.63 mmol) was then added dropwise with rapid stirring. Upon addition of hydrogen peroxide, the previously opaque white solution immediately turned yellow, and over the course of approximately thirty minutes became transparent as the peroxotitanic acid sol formed. The solution was stirred for one hour after the addition of hydrogen peroxide, before being heated to 100 °C with stirring. Heating was maintained for twenty-four hours, during which the transparent yellow solution produced an opaque white or white-yellow solid.
The precipitate was removed from solution by centrifugation (10 min at 5000 rpm) and washed several times with water. For those materials modified by tetrabutylammonium salts, the precipitate was then washed twice with acetonitrile to remove excess tetrabutylammonium salt, followed by a further two washes with water. Finally, products were dried under vacuum to remove remaining solvent.
3.2 Characterisation
Dried products were analysed by powder X-ray diffraction (PXRD) at the Australian Synchrotron. Samples were prepared by packing finely ground dried TiO2 powder in 0.3 mm quartz glass capillary tubes (ultrasound was used to pack the powder down to the bottom of the tubes). The capillary tubes were irradiated for 180 seconds and continuously rotated during the measurement. Data were collected with a Mythen detector spanning 80° in 2θ. Analysis was performed using Topas 4.2 (Bruker ASX). A Rietveld refinement of a NIST standard LaB6 660b was used to determine the incident photons wavelength (0.688650 Å) and instrument profile parameters. A Chebyshev 5th degree polynomial background was used. Structural files of anatase, rutile and brookite were fitted to the PXRD patterns, allowing refinement of crystallite size (Lorentzian and Gaussian), strain (Lorentzian and Gaussian), Ti occupancy, thermal parameters, scale, preferential orientation and lattice parameters. In the case of anisotropic crystallites displaying preferential orientation, two phases of the same lattice parameters, thermal parameters and Ti occupancies were fitted with independently refining degrees of preferential orientation and crystallite size. Crystallite sizes were determined by the average weighted breath of a Double-Voigt fit.36Surface area measurements were performed by BET analysis of N2 adsorption on the samples, using a Micrometrics ASAP2010. Samples were degassed for three hours at 300 °C under 3 μm Hg of vacuum.
Scanning electron microscopy (SEM) images were obtained using a Jeol 7000F FE-SEM. Transmission electron microscopy (TEM) images were obtained using a Philips CM-200 TEM. Samples were suspended in ethanol with the help of sonication, and deposited onto 300 mesh Cu TEM grids coated in Formvar carbon. HRTEM image processing was performed using the software package ImageJ. Dynamic light scattering (DLS) and zeta potential measurements were performed using a Microtrac Zetatrac dynamic light scattering instrument.
The materials were analysed by X-ray photoelectron spectroscopy (XPS) and near-edge X-ray absorption fine-edge structure (NEXAFS) on the soft X-ray beamline at the Australian Synchrotron.41 Samples were dispersed in ethanol and drop-coated onto a silicon wafer prior to measurement.
All XPS measurements were performed using an excitation energy of 825 eV and a pass energy of 10 eV. Data was collected using a SPECS Phoibos 150 hemispherical energy analyser. The background was fitted using a Shirley approximation for all peaks, with pre- and post-peak background simulated using a simple straight-line equation. Binding energies were normalised using the adventitious carbon peak located at 285 eV (assumed to be the most intense C peak when more than one was observed), and intensities were normalised against titanium. In all cases, energy correction was minimal, indicating little to no charging of the material.
NEXAFS was carried out using the same apparatus on the Ti L2,3 edge, with an excitation energy ranging from 450 to 485 eV and a pass energy of 50 eV. Total and Auger Electron Yield (TEY and AEY) as well as Fluorescence Yield (FY) spectra were collected for all samples.
4 Conclusions
Titanium dioxide nanoparticles were synthesised by thermal degradation of peroxotitanic acid in the presence of a number of non-toxic, fluoride-containing, surface-modifying agents (SMAs). The chemical nature of both the anion and cation comprising each SMA affects the properties of the synthesised titania. Overall, the particles were highly crystalline with a high Ti3+ population and residual Ti–F species at the surface. Additional fluoride-containing species were observed on the surface of the particles: treatment with KOH solution was found to remove these species, chemisorbed Ti–F proved surprisingly resistant to removal via this method.The choice of SMA was found to influence particle shape and size. F−- and BF4−-containing SMAs resulted in the formation of monodisperse, ovoid nanoparticles with exposed (010) and (101) faces, while PF6−-containing SMAs systematically favoured formation of much smaller TiO2 particles, resulting in an increased surface area.
Synchrotron XPS and NEXAFS studies provide the first evidence that surface-bound Ti3+ content can be systematically controlled by the choice of SMA. These techniques also reveal two novel trends on the effect of both cations and anions on the abundance of Ti3+ (i.e. F− > BF4− > PF6−; NBu4+ > NH4+). We expect that these properties will lead to increased visible-light activity for the materials due to the creation of intra-band energy levels.
Acknowledgements
The authors acknowledge Mark Jeremy and Mighty River Power for the use of the Nanotrac Zetatrac dynamic light scattering apparatus, and the Australian Synchrotron and NZ Synchrotron Group for synchrotron access. This research was undertaken on the soft X-ray and powder X-ray diffraction beamlines at the Australian Synchrotron, Victoria, Australia. The authors are grateful to Associate Professor Gregory F. Metha from the University of Adelaide for discussions relating to XPS peak and background fitting. We thank Professor Milo Kral for access to the electron microscopy facility at the University of Canterbury. Funding was provided by the MacDiarmid Institute, the University of Canterbury, the Australian Synchrotron (grants AS121/SXRFI/4641a, AS121/SXR/4641b and AS122/SXR/5065), the New Zealand Synchrotron Group, and the Ministry for Business, Innovation and Employment New Zealand (contract C05X1207).References
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- X. Hu, G. Li and J. C. Yu, Langmuir, 2010, 26, 3031–3039 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- A. Fujishima, X. Zhang and D. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- G. Liu, L. Wang, H. G. Yang, H.-M. Cheng and G. Q. M. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- H. Sun, S. Wang, H. M. Ang, M. O. Tadé and Q. Li, Chem. Eng. J., 2010, 162, 437–447 CrossRef CAS PubMed.
- H. Sun, Y. Bai, Y. Cheng, W. Jin and N. Xu, Ind. Eng. Chem. Res., 2006, 45, 4971–4976 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H.-M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- S. Ahmed, M. G. Rasul, R. Brown and M. A. Hashib, J. Environ. Manage., 2011, 92, 311–330 CrossRef CAS PubMed.
- M. A. Henderson, Surf. Sci. Rep., 2011, 66, 185–297 CrossRef CAS PubMed.
- H. Park and W. Choi, J. Phys. Chem. B, 2004, 108, 4086–4093 CrossRef CAS.
- M. V. Dozzi, B. Ohtani and E. Selli, Phys. Chem. Chem. Phys., 2011, 13, 18217–18227 RSC.
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H.-M. Cheng and G. Q. M. Lu, J. Am. Chem. Soc., 2009, 131, 4078–4083 CrossRef CAS PubMed.
- X. Han, Q. Kuang, M. Jin, Z. Xie and L. Zheng, J. Am. Chem. Soc., 2009, 131, 3152–3153 CrossRef CAS PubMed.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, H. Lu, L. Wang, G. Q. Lu and H.-M. Cheng, J. Phys. Chem. C, 2009, 113, 21784–21788 CAS.
- J. Feng, M. Yin, Z. Wang, S. Yan, L. Wan, Z. Li and Z. Zou, CrystEngComm, 2010, 12, 3425 RSC.
- X. H. Yang, Z. Li, G. Liu, J. Xing, C. Sun, H. G. Yang and C. Li, CrystEngComm, 2011, 13, 1378–1383 RSC.
- A. S. Ichimura, B. M. Mack, S. M. Usmani and D. G. Mars, Chem. Mater., 2012, 24, 2324–2329 CrossRef CAS PubMed.
- N. Liu, Y. Zhao, X. Wang, H. Peng and G. Li, Mater. Lett., 2013, 102–103, 53–55 CAS.
- L. Wu, B. X. Yang, X. H. Yang, Z. G. Chen, Z. Li, H. J. Zhao, X.-Q. Gong and H. G. Yang, CrystEngComm, 2013, 15, 3252 RSC.
- D. Zhang, G. Li, X. Yang and J. C. Yu, Chem. Commun., 2009, 4381–4383 RSC.
- J. Yu, Q. Xiang and J. R. S. Mann, CrystEngComm, 2010, 12, 872–879 RSC.
- C. Cai, J. Wang, F. Cao, H. Li and J. Zhu, Chin. J. Catal., 2011, 32, 862 CrossRef CAS.
- Z. Lai, F. Peng, Y. Wang, H. Wang, H. Yu, P. Liu and H. Zhao, J. Mater. Chem., 2012, 22, 23906 RSC.
- K. Lv, J. Yu, J. Fana and M. Jaroniec, CrystEngComm, 2011, 13, 7044–7048 RSC.
- X. Zhou, F. Peng, H. Wang, H. Yu and Y. Fang, Chem. Commun., 2012, 48, 600–602 RSC.
- J. Zhu, S. Wang, Z. Bian, S. Xie, C. Cai, J. Wang, H. Yang and H. Li, CrystEngComm, 2010, 12, 2219–2224 RSC.
- D. Wu, Z. Gao, F. Xu, J. Chang, S. Gao and K. Jiang, CrystEngComm, 2012, 15, 516 RSC.
- W. Wang, C. Lu, Y. Ni and Z. Xu, CrystEngComm, 2013, 15, 2537 RSC.
- L. Zhou, J. Chen, C. Ji, L. Zhou and P. O'Brien, CrystEngComm, 2013, 15, 5012 RSC.
- X. Ding, H. Ruan, C. Zheng, J. Yang and M. Wei, CrystEngComm, 2013, 15, 3040 RSC.
- W. Wang, C. Lu, Y. Ni, F. Peng and Z. Xu, Appl. Surf. Sci., 2012, 1–5 CrossRef PubMed.
- J. C. Yu, J. Yu, W. Ho, Z. Jiang and L. Zhang, Chem. Mater., 2002, 174, 3808–3816 CrossRef.
- D. Balzar, Microstructure Analysis from Diffraction, International Union of Crystallography, 1999 Search PubMed.
- B. A. Morales, O. Novaro, T. Lopez, E. Sanchez and R. Gomez, J. Mater. Res., 1995, 10, 2788–2796 CrossRef CAS.
- C. Ribeiro, C. Vila, D. B. Stroppa, V. R. Mastelaro, J. Bettini, E. Longo and E. R. Leite, J. Phys. Chem. C, 2007, 111, 5871–5875 CAS.
- W.-J. Li, E.-W. Shi and Z.-W. Yin, J. Cryst. Growth, 2000, 208, 546–554 CrossRef CAS.
- J. Pan, G. Liu, G. Q. M. Lu and H.-M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- B. C. C. Cowie, A. Tadic and L. Thomsen, AIP Conf. Proc., 2010, 1234, 307–310 CrossRef PubMed.
- Z. He, Q. Cai, F. Hong, Z. Jiang, J. Chen and S. Song, Ind. Eng. Chem. Res., 2012, 51, 5662–5668 CrossRef CAS.
- Q. Wang, C. Chen, D. Zhao, W. Ma and J. Zhao, Langmuir, 2008, 24, 7338–7345 CrossRef CAS PubMed.
- T. K. Le, D. Flahaut, H. Martinez, T. Pigot, H. K. H. Nguyen and T. K. X. Huynh, Appl. Catal., B, 2014, 144, 1–11 CrossRef CAS PubMed.
- D. Li, H. Haneda, S. Hishita and N. Ohashi, Chem. Mater., 2005, 17, 2588–2595 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Physical Electronics Limited, Minnesota, 1995 Search PubMed.
- Q. Xiang, K. Lv and J. Yua, Appl. Catal., B, 2010, 96, 557–564 CrossRef CAS PubMed.
- Y. Kim, B. J. Yoo, R. Vittal, Y. Lee, N.-G. Park and K.-J. Kim, J. Power Sources, 2008, 175, 914–919 CrossRef CAS PubMed.
- X. Zhang, H. Tian, X. Wang, G. Xue, Z. Tian, J. Zhang, S. Yuan, T. Yu and Z. Zou, Mater. Lett., 2013, 100, 51–53 CrossRef CAS PubMed.
- S. Song, J. Tu, Z. He, F. Hong, W. Liu and J. Chen, Appl. Catal., A, 2010, 378, 169–174 CrossRef CAS PubMed.
- S. A. Tenney, B. A. Cagg, M. S. Levine, W. He, K. Manandhar and D. A. Chen, Surf. Sci., 2012, 606, 1233–1243 CrossRef CAS PubMed.
- G. Liu, J. A. Rodriguez, J. Hrbek, B. T. Long and D. A. Chen, J. Mol. Catal. A: Chem., 2003, 202, 215–227 CrossRef CAS.
- G. G. Fuentes, E. Elizalde, F. Yubero and J. M. Sanz, Surf. Interface Anal., 2002, 33, 230–237 CrossRef CAS.
- T. Kako, N. Umezawa, K. Xie and J. Ye, J. Mater. Sci., 2013, 48, 108–114 CrossRef CAS.
- F. Zuo, L. Wang, T. Wu, Z. Zhang, D. Borchardt and P. Feng, J. Am. Chem. Soc., 2010, 132, 11856–11857 CrossRef CAS PubMed.
- J. P. Crocombette and F. Jollet, J. Phys.: Condens. Matter, 1994, 6, 10811–10821 CrossRef CAS.
- R. Ruus, A. Kikas, A. Saar, O. A. Ausmees, E. Ndmmiste, J. Aarik, A. Aidla, T. Uustare and I. Martinson, Solid State Commun., 1997, 104, 199–203 CrossRef CAS.
- M. Cheynet, S. Pokrant, S. Irsen and P. Krüger, Ultramicroscopy, 2010, 110, 1046–1053 CrossRef CAS PubMed.
- G. S. Henderson, X. Liu and M. E. Fleet, Phys. Chem. Miner., 2002, 29, 32–42 CrossRef CAS.
- S. J. Stewart, M. Fernández-García, C. Belver, B. S. Mun and F. G. Requejo, J. Phys. Chem. B, 2006, 110, 16482–16486 CrossRef CAS PubMed.
- J. Stöhr, NEXAFS Spectroscopy, Springer, Berlin, 1st edn, 2003 Search PubMed.
- F. De Groot, M. O. Figueiredo, M. J. Basto, M. Abbate, H. Petersen and J. C. Fuggle, Phys. Chem. Miner., 1992, 19, 140–147 CrossRef CAS.
- Z. Miao, Z. Liu, K. Ding, B. Han, S. Miao and G. An, Nanotechnology, 2007, 18, 125605 CrossRef.
- P. J. Dyson and T. J. Geldbach, Metal Catalysed Reactions in Ionic Liquids, Springer, Dordrecht, 2005 Search PubMed.
- D. S. Jacob, L. Bitton, J. Grinblat, I. Felner, Y. Koltypin and A. Gedanken, Chem. Mater., 2006, 18, 3162–3168 CrossRef CAS.
- N. Sasirekha, B. Rajesh and Y.-W. Chen, Thin Solid Films, 2009, 518, 43–48 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures for characterisation of materials. See DOI: 10.1039/c3ra47652a |
| This journal is © The Royal Society of Chemistry 2014 |