A single rapid route for the synthesis of reduced graphene oxide with antibacterial activities†
Suela Kellici*a,
John Acorda,
Jeremy Ballb,
Haricharan S. Reehalb,
David Morganc and
Basudeb Saha*a
aDepartment of Applied Sciences, Faculty of Engineering, Science and the Built Environment, London South Bank University, 103 Borough Road, London, SE1 0AA, UK. E-mail: b.saha@lsbu.ac.uk; kellicis@lsbu.ac.uk
bDepartment of Engineering and Design, Faculty of Engineering, Science and the Built Environment, London South Bank University, 103 Borough Road, London, SE1 0AA, UK
cCardiff Catalysis Institute, School of Chemistry, Cardiff University, Park Place, Cardiff, CF10 3AT, UK
First published on 12th March 2014
Abstract
An innovative approach is employed for synthesising reduced graphene oxide with antibacterial properties via utilisation of continuous supercritical water (in alkaline medium) which enabled reduction of graphene oxide in a single rapid route and thus eliminating hazardous conventional chemicals.
Graphene, a single sheet of hexagonally arrayed sp2-bonded carbon atoms, is one of the most promising materials in nanotechnology.1 Its unique physical, chemical and mechanical properties including a very high surface area and easy surface modification allow preparation of nanocomposite materials with novel properties and characteristics.2 Additionally, graphene based materials have been reported to have superior antibacterial properties with minimal or no cytotoxicity to human and animal cells.3 As such these materials may be employed for biomedical applications.4
One of the most common synthetic approaches for graphene production involves the initial oxidation of graphite to graphite oxide, followed by chemical exfoliation of graphite oxide to graphene oxide sheets (GO).‡5 However, the reduction of graphite oxide to graphene (rGO) is required to recover the conjugated network as well as electrical conductivity, a process which mainly involves the utilisation of toxic and very unstable (potentially explosive) reducing agents such as hydrazine which in turn also leave trace residues inducing deterioration in the performance of the end material. In this context, the preparation of high quality 2D graphene sheets through green synthetic routes is the most crucial and desirable step, since the presence of defects will influence the properties and consequently its applications. For preparation of metal oxides, hydrothermal syntheses in superheated or supercritical water can offer many advantages over conventional preparative methods.6–9 Notably, lower synthesis temperatures and fewer processing steps are required. There are currently very few scientific reports utilising hydrothermal synthetic procedures for graphene reduction.8,10 Also, the current hydrothermal syntheses are conducted in batch reactors, which are time consuming and give little control over product properties. Continuous hydrothermal flow synthesis (CHFS) reactors offer many advantages as they have independent control over reaction parameters (e.g. temperature and pressure) and hence particle properties. Additionally, it offers significantly faster reaction times.11 The CHFS process involves mixing a flow of supercritical water with a flow of aqueous metal salt(s) to give rapid precipitation and controlled growth of nanoparticles at a defined mixing zone in a continuous manner (Fig. 1).§ A key feature of this process is the way in which the properties of water (such as density, diffusivity and dielectric constant) change dramatically around the critical T, P (374 °C, 22.1 MPa) leading to its use as an exotic, highly controllable reaction solvent/medium.12 Additionally, CHFS process can be adapted (as shown by previously published work)11 to high throughput methods for the synthesis of materials in a relatively short timeframe, thus, allowing for the rapid production and evaluation of materials. Herein, we report a new approach using supercritical water to make advanced graphene based functional materials with antibacterial properties. In our process, the aqueous solution of GO was pumped to mix with a flow of KOH at room temperature at a T-junction (‘T’ in Fig. 1). This mixture was then brought into contact with supercritical water in counter-current reactor (R), whereupon reduction of graphene oxide (rGO) occurred. Transmission Electron Microscopy (TEM) images of GO and rGO are shown in Fig. 2a and b, respectively. Scanning Electron Microscopy (SEM) images of graphene shown in Fig. 2c revealed graphene sheets with wrinkles. However, the SEM images of reduced graphene oxide prepared via hydrothermal method showed a more defined flake like morphology. This was also confirmed by Atomic Force Microscopy (AFM) images of graphene and reduced graphene oxide as shown in Fig. 2e and f, respectively.
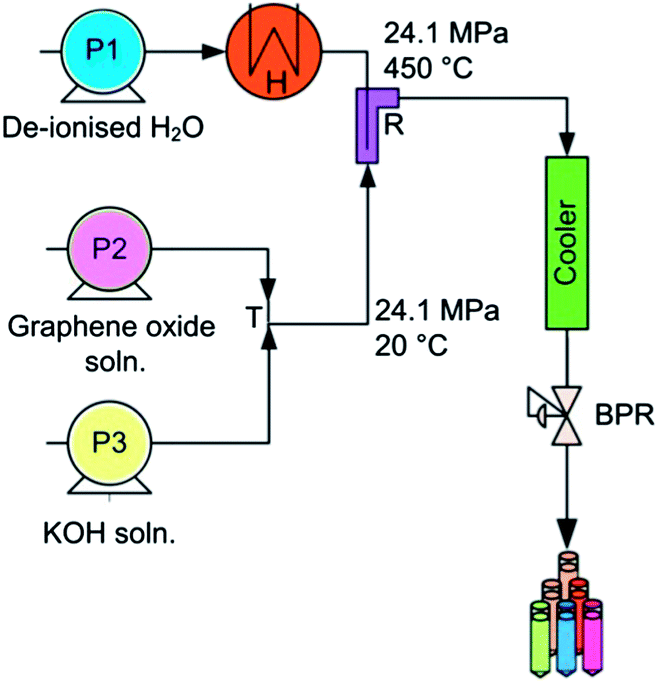 | ||
| Fig. 1 Schematic representation of CHFS reactor utilised for the synthesis of reduced graphene oxide. Key: P = HPLC pump, H = heater, T = tee junction, R = reactor, BPR = back pressure regulator. | ||
 | ||
| Fig. 2 Transmission Electron Microscopy (TEM) images of (a) graphene and (b) reduced graphene oxide materials. Scanning Electron Microscopy images (SEM) of (c) graphene oxide and (d) reduced graphene oxide and Atomic Force Microscopy (AFM) images of (e) graphene oxide and (f) reduced graphene oxide synthesised via continuous hydrothermal process. | ||
Raman spectroscopy (Fig. 3(I)) was employed to study the effect of the hydrothermal process and the reduction of graphene oxide on the carbon structure of the graphene materials. The characteristic D band (located at ca. 1350 cm−1) and G bands (1580 cm−1) are attributed to the local defects/disorders (found at the edges of graphene sheets) and the sp2 graphitized structure, respectively. Thus, smaller ID/IG peak intensity ratios are assigned to lower defects/disorders. Raman spectra show an intensity ratio of ID/IG of 0.71 and 0.76 for rGO and GO, respectively. This indicates a lower defect levels for rGO samples made hydrothermally. The crystallinity of the nano-powders was assessed by X-ray powder diffraction (XRD) and is shown in Fig. 3(II). GO (Fig. 3(II)c) exhibits an intense peak located at a low angle region, ca. 2θ = 11.48°, corresponding to d-spacing of 0.77 nm along the (002) orientation. XRD pattern of rGO revealed a broad peak (002) at 2θ = 25.32° corresponding to d-spacing of 0.38, indicating the formation of highly graphitized material.
 | ||
| Fig. 3 (I) Raman spectra of (a) graphene oxide and (b) reduced graphene oxide materials. (II) X-ray powder diffraction pattern of (c) graphene oxide and (d) reduced graphene oxide synthesised via CHFS process. | ||
To investigate the changes in the chemical states of the GO and rGO materials, X-ray photoelectron spectroscopy (XPS) was employed, the spectra of which are shown in Fig. 4. For comparison, reduced graphene oxide was made conventionally¶15 (rGOc) and the XPS spectrum was also recorded (see Fig. S1, ESI†).|| The deconvoluted C(1s) XPS spectra of the GO shows a considerable degree of oxidation confirmed by the presence of carbon atoms in functional groups of epoxide, hydroxyl and carboxyl. For rGOc there is a large increase in the species at 286.6 eV (>C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the development of a strong peak at 290.4 eV (–COOH). It is interesting to note that other researchers assign the species around 290.5 eV to the π–π* transition indicating some restoration of the aromatic structure in rGO, whilst this is true based on their XPS results, the intensity (and therefore concentration of the species) of the peak here does not support this as the single reasoning, instead we observe definitive formation of surface acid groups (–COOH). However, for rGO material made hydrothermally there is a significant reduction in peak intensities of the oxygen-containing functional groups. This confirms that continuous hydrothermal process is effective in dehydrating/reducing GO.
O) and the development of a strong peak at 290.4 eV (–COOH). It is interesting to note that other researchers assign the species around 290.5 eV to the π–π* transition indicating some restoration of the aromatic structure in rGO, whilst this is true based on their XPS results, the intensity (and therefore concentration of the species) of the peak here does not support this as the single reasoning, instead we observe definitive formation of surface acid groups (–COOH). However, for rGO material made hydrothermally there is a significant reduction in peak intensities of the oxygen-containing functional groups. This confirms that continuous hydrothermal process is effective in dehydrating/reducing GO.
 | ||
| Fig. 4 XPS spectra of (a) graphene oxide and (b) reduced graphene oxide. | ||
Furthermore, the deoxidation is confirmed by FTIR (Fig. 5), which revealed that strongest absorption band assigned to >CO/COOH group (1713 cm−1) reduced significantly and the vibration band of epoxy group (1032 cm−1) as well as the strong band located at 1151 cm−1 (C–OH stretching vibrations) disappeared. Also, elemental analysis (Table S1, ESI†) revealed an increase in C/O atomic ratio in rGO compared to the starting GO.
 | ||
| Fig. 5 FT-IR spectra of (a) graphene oxide and (b) reduced graphene oxide. | ||
The antibacterial properties of GO and rGO were tested using a microdilution method16 against the gram negative bacterium Escherichia coli (E. coli)** to determine the minimum inhibitory concentration (MIC). The MIC for reduced graphene oxide produced via the hydrothermal flow method (rGOht) was the same as that for reduced graphene oxide produced via the conventional method (rGOc) as presented in Table 1. However, we also found that graphene oxide (GO) did not inhibit the growth of E. coli at any concentrations tested (up to 10 mg mL−1).
| Compound | MIC (μg mL−1) |
|---|---|
| GO | Unable to determine |
| rGOc | 125 |
| rGOht | 125 |
To determine if rGO displayed bactericidal activity, E. coli were incubated for 6 hours with 1×, 2× and 4× the MIC for rGOht, 1× the MIC for rGOc and 500 μg mL−1 GO. Samples were taken every 90 minutes and the number of surviving colonies enumerated (Fig. 6).
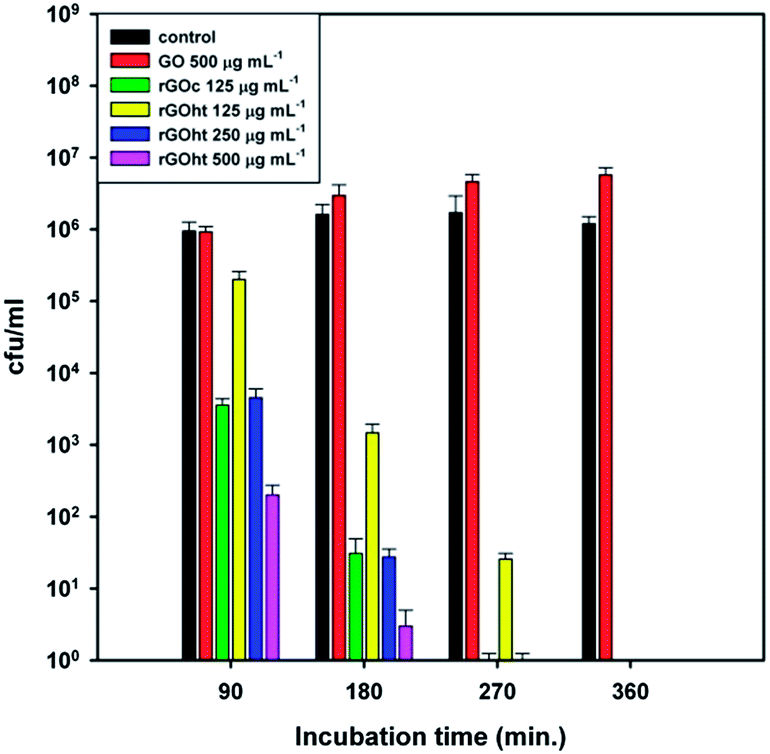 | ||
| Fig. 6 Cell viability measurements of Escherichia coli after incubation at 37 °C with GO, rGOc and rGOht at various concentrations and incubation times. | ||
The observation that rGOht takes longer than rGOc to reduce bacterial numbers to undetectable levels may be explained by the differences seen between these compounds when compared by XPS (Fig. S1, ESI†) and therefore may indicate that the –COOH groups seen in rGOc are important for antibacterial activity. However the enhanced antibacterial activity of rGOht compared to GO agree with previous research findings.3,4 It is also worth noting that GO did not kill E. coli at the concentration tested, and may instead have served to modestly enhance bacterial growth, this is in agreement with the observations previously reported by Ruiz, et al.17
Conclusions
In conclusion, a rapid continuous hydrothermal flow route was employed for the synthesis of rGO with antibacterial properties. The versatility of this synthetic route should allow the rapid synthesis of more graphene based nanomaterials. In fact, our unpublished results are highly promising in this regard, the detailed work will be reported in due course.Acknowledgements
We acknowledge Mr. Steve Jones (LSBU) for his tremendous technical support with the CHFS reactor development. Dr. Clive Steele and Mr. Peter Adams (LSBU) are thanked for the technical support. Dr. Zofia Luklinksa (QMUL) is acknowledged for her help with the EM images. Dr. Nicholas Power (LSBU) is thanked for useful discussions.Notes and references
- (a) A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed; (b) C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS PubMed; (c) A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- (a) D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC; (b) D. Li and R. B. Kaner, Science, 2008, 320, 1170 CrossRef CAS PubMed.
- (a) W. Hu, C. Peng, W. Luo, M. Lv, X. Li, D. Li, Q. Huang and C. Fan, ACS Nano, 2010, 4, 4317 CrossRef CAS PubMed; (b) Q. Bao, D. Zhang and P. Qi, J. Colloid Interface Sci., 2011, 360, 463 CrossRef CAS PubMed.
- (a) O. Akhavan and E. Ghaderi, ACS Nano, 2010, 4, 5731 CrossRef CAS PubMed; (b) J. Shen, M. Shi, N. Li, B. Yan, H. Ma, Y. Hu and M. Ye, Nano Res., 2010, 3, 339 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217 CrossRef CAS PubMed.
- J. A. Darr and M. Poliakoff, Chem. Rev., 1999, 99, 495 CrossRef CAS PubMed.
- A. A. Chaudhry, S. Haque, S. Kellici, P. Boldrin, I. Rehman, F. A. Khalid and J. A. Darr, Chem. Commun., 2006,(21), 2286 RSC.
- Y. Zhou, Q. Bao, L. A. L. Tang, Y. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950 CrossRef CAS.
- K. Sue, K. Kimura, M. Yamamoto and K. Arai, Mater. Lett., 2004, 58, 3350 CrossRef CAS.
- J. Shen, M. Shi, B. Yan, H. Ma, N. Li and M. Ye, J. Mater. Chem., 2011, 21, 7795 RSC.
- (a) S. Kellici, K. Gong, T. Lin, S. Brown, R. J. H. Clark, M. Vickers, J. K. Cockcroft, V. Middelkoop, P. Barnes, J. M. Perkins, C. J. Tighe and J. A. Darr, Philos. Trans. R. Soc., A, 2010, 368, 4331 CrossRef CAS PubMed; (b) T. Lin, S. Kellici, K. Gong, K. Thompson, J. R. G. Evans, X. Wang and J. A. Darr, J. Comb. Chem., 2010, 12, 383 CrossRef CAS PubMed; (c) X. Weng, J. K. Cockcroft, G. Hyett, M. Vickers, P. Boldrin, C. C. Tang, S. P. Thompson, J. E. Parker, J. C. Knowles, I. Rehman, I. Parkin, J. R. G. Evans and J. A. Darr, J. Comb. Chem., 2009, 11, 829 CrossRef CAS PubMed.
- (a) Z. Zhang, S. Brown, J. B. M. Goodall, X. Weng, K. Thompson, K. Gong, S. Kellici, R. J. H. Clark, J. R. G. Evans and J. A. Darr, J. Alloys Compd., 2009, 476, 451 CrossRef CAS; (b) X. Weng, P. Boldrin, I. Abrahams, S. J. Skinner, S. Kellici and J. A. Darr, J. Solid State Chem., 2008, 181, 1123 CrossRef CAS.
- (a) Y. Hakuta, T. Adschiri, T. Suzuki, T. Chida, K. Seino and K. Arai, J. Am. Ceram. Soc., 1998, 81, 2461 CrossRef CAS; (b) A. Cabanas and M. Poliakoff, J. Mater. Chem., 2001, 11, 1408 RSC.
- E. Lester, P. Blood, J. Denyer, D. Giddings, B. Azzopardi and M. Poliakoff, J. Supercrit. Fluids, 2006, 37, 209 CrossRef CAS.
- J. Tian, S. Liu, Y. Zhang, H. Li, L. Wang, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Inorg. Chem., 2012, 51, 4742 CrossRef CAS PubMed.
- D. Amsterdam, Susceptibility testing of antimicrobials in liquid media, in Antibiotics in Laboratory Medicine, ed. V. Lorian, Williams & Wilkins, Baltimore MD, 4th edn, 1996, p. 52 Search PubMed.
- O. N. Ruiz, K. A. S. Fernando, B. Wang, N. A. Brown, P. G. Luo, N. D. McNamara, M. Vangsness, Y. Sun and C. E. Bunker, ACS Nano, 2011, 5(10), 8100 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS elemental analysis, XPS spectra for rGOc sample made conventionally. See DOI: 10.1039/c3ra47573e |
| ‡ Experimental methods: GO was synthesised using a modified Hummers method from graphite.13 |
| § CHFS experiments were conducted using a flow reactor design to that reported previously.7,12a The system consists of three HPLC pumps used for the delivery of aqueous solution of reagents. The tubing and fittings were 1/8′′ 316SS Swagelok, except the counter-current reactor and the cooler, which were constructed using 1/4 inch fittings. Pump 1 (Gilson 307 fitted with 25 mL pump head) was utilised for delivering water through a custom made pre-heater (H) at a flow rate of 20 mL min−1. Pump 2 and Pump 3 (Varian Pro Star 210 fitted with 5 mL pump head) were employed for pumping aqueous GO solution and KOH at a flow rate of 5 mL min−1. In a typical experiment, a pre-sonicated (30 min) aqueous solution of GO (4 μg mL−1) was pumped to meet a flow of KOH (0.2 M) at a T-junction (T in Fig. 1). This mixture then meet superheated water (450 °C, 24.1 MPa) inside an in-house built 1/4 inch counter-current mixer14 (R in Fig. 1) whereupon the reduction of graphene oxide occurred in a continuous matter. The aqueous suspension was cooled through a vertical cooler and the slurries were collected from the exit of the back pressure regulator (BPR in Fig. 1). After collection, the particles were centrifuged and washed with water twice. The solids were freeze-dried. |
| ¶ A control reaction studying the effect of the treatment on the de-oxygenation of graphene oxides (GO) using a conventional method was also conducted.15 |
|| Equipment and techniques: Freeze-drying was performed using a Heto PowderDry PL 3000. Raman spectra were collected using a Renishaw 1000 using an excitation source of a 50 mW 488 nm Stellar-pro laser. AFM images were recorded in the contact mode with a Nanoscope IIIa (Digital Instruments). A JEOL 2010 TEM (200 kV accelerating voltage) was used for generating images of particles. XRD data were collected on a Bruker D2 Phaser X-ray diffractometer using Cu-Kα radiation (l = 0.15418 nm) for 10 h, range 10–60 2θ with a step size of 0.02. XPS measurements were performed using a Kratos Axis ultra DLD photoelectron spectrometer utilising monochromatic Alka source operating at 144 W. Samples were mounted using conductive carbon tape. Survey and narrow scans were performed at constant pass energies of 160 and 40 eV respectively. The base pressure of the system is ca. 1 × 10−9 Torr, rising to ca. 4 × 10−9 Torr under analysis of these samples. FT-IR spectra was recorded using a Nicolet Avatar 370DTGS spectrometer fitted with a Smart Orbit accessory (diamond 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–200 cm−1). 000–200 cm−1). |
| ** The antibacterial activity of graphene oxide or reduced graphene oxides was tested using the gram negative bacterium E. coli (ATCC 47076). To determine MIC values an overnight culture of E. coli was diluted into fresh LB media (10 g L−1 tryptone, 5 g L−1 yeast extract, 5 g L−1 NaCl) to give a concentration of approximately 106 cfu mL−1. The culture was aseptically transferred to a microtiter plate and the compounds being tested for antimicrobial activity were added at various concentrations ranging from 10 mg mL−1 in two-fold serial dilutions to 7.8 μg mL−1. The plate was incubated overnight at 37 °C and the concentration of compound that resulted in visible inhibition of growth was determined the following day. Bactericidal activity was measured by diluting an overnight culture of E. coli into phosphate buffered saline to a final concentration of 106 cfu mL−1 and incubating in the presence of GO, rGOc and rGOht at various concentrations for 6 h at 37 °C with constant agitation. Samples were taken every 90 minutes, serially diluted, and 100 μL spread on LB agar plates (as per LB media but with 1% w/v agar) which were then incubated overnight at 37 °C. Colonies were counted the following day. All experiments were performed in triplicate and repeated twice. The results shown are the mean of the values obtained for each time point, and error bars represent standard deviation. |
| This journal is © The Royal Society of Chemistry 2014 |