
Synthesis of pyrrolidine iminosugars, (−)-lentiginosine, (−)-swainsonine and their 8a-epimers from d-glycals†
Abstract
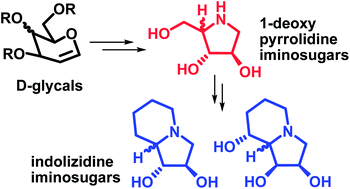
Synthesis of pyrrolidine iminosugars has been described from D-glycals via dihydroxylation, oxidative cleavage and double nucleophilic displacement as the key steps. The pyrrolidines obtained have been utilized for the synthesis of important bicyclic iminosugars, viz. (−)-lentiginosine and (−)-swainsonine and their 8a-epimers, which are known to be glycosidase inhibitors.
 Please wait while we load your content...
Please wait while we load your content...

