Encapsulation of Au nanoparticles with well-crystallized anatase TiO2 mesoporous hollow spheres for increased thermal stability
Zewu Zhang,
Yuming Zhou*,
Yiwei Zhang,
Sanming Xiang,
Shijian Zhou and
Xiaoli Sheng
School of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, P.R.China. E-mail: ymzhou@seu.edu.cn; Fax: +86 25 52090617; Tel: +86 25 52090617
First published on 8th January 2014
Abstract
Uniform Auencap/TiO2 hollow microspheres, in which sub-10 nm Au nanoparticles are coated with a mesoporous anatase TiO2 shell, are prepared by a protected-calcinating process. The method involves the preparation of SiO2@Au@TiO2 colloidal composites, sequential deposition of carbon and then SiO2 layers through solvothermal and sol–gel processes, crystallization of TiO2 by calcination and finally etching of the inner and outer silica layers to produce the hollow structure. The protected crystallization process suppresses the excessive growth of TiO2 and eventually produces mesoporous anatase shells with high surface area. Additionally, by simply controlling the crystallization temperature, it can be convenient to tune the porosity and crystallinity of the TiO2 shell, meanwhile maintaining the small size of Au nanoparticles. When used as the catalyst for the reduction of 4-nitrophenol, the synthesized Auencap/TiO2 catalysts exhibit significantly enhanced catalytic performance. Moreover, the obtained Auencap/TiO2 hollow spheres show a superior thermal stability, as it resists sintering during the additional calcination at 500 °C, whereas the sample prepared by deposited Au nanoparticles on commercial P25(Au/P25) was found to sinter severely.
1. Introduction
The advantages of nanosized noble metal particles in heterogeneous catalysis have been widely recognized in the past few years.1,2 At the nanoscale, the particles exhibit fascinating physical, chemical and biological properties that are often different to the bulk counterparts, which bestows them with high potentials for catalysis, sensor, biomedicine and so on.3–5 As one of the most investigated nanoparticle systems, gold nanoparticles (Au NPs) have been successfully applied in various catalytic reactions including low-temperature CO oxidation,6 photodegradation of organic pollutants,7 Fischer–Tropsch reaction8 and so forth.9 Recent advances in colloid chemistry have enabled us to prepare Au NPs with tunable size, shape and composition.10,11However, challenges still exist. A bottleneck problem for Au nanocatalysts is their lack of stability at realistic technical conditions.6 It is well known that Au NPs with small sizes are usually tend to diffuse, coalesce and sinter under the harsh conditions, and that leads to loss of the unique properties registered in the original nanoparticles.12 To solve the problem of migration and aggregation, many strategies have been carried out. Among them, encapsulating the nanoparticles in a porous oxide shell have been proposed as one of the effective means to improve the thermal stability of the metal nanocatalysts.13–15 Using this strategy, many catalysts with core–shell or yolk–shell structures have been prepared, in which the individual gold particle is isolated by the oxide shell (MOx). The shell restrict the migration of the neighboring gold particles, and therefore the particle can be prevented from sintering. Meanwhile, construction pore in the oxide shell may allow the chemical species to reach the buried catalytic particles and therefore the reaction activity is improved.16,17 Nevertheless, in this architecture, usually named nanoreactors,18 only one metal particle exists in each composite particle. The little gold contents in the catalysts may affect their catalytic performance. Actually, many obtained MOx coated Au nanocatalysts shows lower activity in comparison with the supported Au/MOx catalysts with the same catalysts dosage.6,19,20
To increase the amount of Au NPs in catalytic particles, people have developed a new construction that combines the supported metal nanoparticles and surface-encapsulation methods.14,21,22 In their systems, the Au loading is increased largely and the catalytic activity is enhanced to some extent. However, the inorganic coating used here is mostly limited to SiO2, for it's easy to synthesize and high-temperature stability.13,23 It is generally believed that the SiO2 is an inert support, which has weak metal–support interaction.24 In many catalytic reactions, SiO2 does not contribute to the catalytic activity, and thus the gold supported on SiO2 catalysts usually possess poor activity.25,26 As a result, the metal oxides are of increasing interest matrix for its synergistic effect in the promotion of catalytic reaction. However, the metal oxides are mainly used as the support to disperse the metal particles still now. In this architecture, the degree of integration between Au NPs and metal oxides is relatively low, which deceases the contact area of the two components. It is well known that the surface encapsulation can significantly increase the interaction of the active metal and the oxide shell. Thus, it is highly desirable to encapsulate the supported Au NPs with metal oxide.
Generally speaking, two methods, “surfactant-templating” and “surface-protected etching”, have been used to synthesis the permeable SiO2 shells.27,28 However, few reports about the preparation of the activated metal oxide porous shell, especially for the crystalline TiO2. One big challenge is that classical calcination schema to crystallization the amorphous TiO2 often leads to significant decrease in the surface area of the TiO2 shells.29 To solve this problem, Yin and us have recently proposed many methods based on “surface-protected calcination” and obtained anatase TiO2 with well-developed crystallinity and favorable mesoporous structure.30,31 These greatly inspire us to develop novel porous anatase TiO2 shell encapsulated Au nanoparticles.
Herein, we report such a gold nanocatalyst composed of porous anatase TiO2 hollow shell with Au NPs embedded in the inner surfaces (Auencap/TiO2, Fig. 1). The structure is different from those of reported Au@TiO2 catalysts for abundant sub-10 nm gold particles in one catalytic particle in our catalyst system.20 The synthetic procedure involves several sequential steps as follows: (1) synthesis of SiO2@Au@TiO2 colloidal composites through sol–gel processes, (2) sequential coating of the colloidal composites with carbon and SiO2 layer, (3) calcination to crystallization of the TiO2 shell, (4) remove the overall silica by etching with NaOH solution. Thanks to the effective metal–TiO2 interaction, the obtained catalysts show superior thermal stability, as the encapsulated Au NPs can resist sintering up to 500 °C in air. Impressively, when these Auencap/TiO2 microspheres are used as the catalyst for reduction of 4-nitrophenol (4-NP), they exhibit considerably enhanced catalytic activity and reusability.
 | ||
| Fig. 1 Schematic illustration of our silica-protected calcination procedure developed for the encapsulation of supported Au nanoparticles with mesoporous anatase TiO2 hollow spheres. | ||
2. Experimental section
Synthesis of SiO2/Au/TiO2
The SiO2/Au/TiO2 were prepared by modified the strategy described by Yin.32 Briefly, spherical silica microspheres, about 200 nm in diameter, were first synthesis by mixing tetraethylorthosilicate (TEOS, 1.72 mL), concentrated ammonia solution (28 wt%, 1.24 mL), ethanol (46 mL) and deionized water (8.6 mL) at room temperature for 6 h, and then functionalized with amino groups by refluxing a mixture of isopropanol (50 mL) and 3-aminopropyl-triethoxysilane (APTES, 1 mL) at 80 °C for 12 h. In a separate reaction, the Au nanoparticles were synthesized by mixing a solution of HAuCl4 (356 μL) with deionized water (75 mL) and sodium citrate solution (40 mg mL−1, 0.55 mL), quickly injecting a sodium borohydride solution (0.1 mol L−1, 4.5 mL) under vigorous stirring. The resulting citrate-stabilized Au nanoparticles, 5 nm in diameter, were then adsorbed on the SiO2–NH2 colloids. The TiO2 layer was deposited by mixing the SiO2–Au colloidal spheres with hydroxypropyl cellulose (HPC, 0.2 g), ethanol (50 mL) and deioned water (0.2 mL). After stirring for 30 min, a mixed solution containing titanium tert-butoxide (TBOT, 2 mL) and 10 mL anhydrous ethanol was slowing injected, and then followed by refluxing at 85 °C for 100 min. The particles were isolated by centrifugal separation, washed with ethanol. To obtain a thicker TiO2 shell, the TiO2 coating process was repeated.Sequential coating with carbon and SiO2
The carbon layer was prepared by a solvothermal reaction. In a typical experiment, the SiO2/Au/TiO2 spheres were mixed with a 20 mL PVP solution (Mw ∼ 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 0.5 g), then centrifuging the mixture to remove unbound polymer and dispersing the solid into a mixture of ethanol, deioned water (5 mL) and glucose (1 g). The mixture was then sealed into a Teflon-lined autoclave. After reaction at 180 °C for 12 h, the carbon coated particles were centrifuged, washed and dispersed in a PVP solution for additional surface modified. Then the suspension was mixed with ethanol (36 mL), water (0.86 mL), aqueous ammonia (0.64 mL) and TEOS (0.86 mL), followed by stirring for 6 h, the SiO2/Au/TiO2/C/SiO2 composite particles were collected by centrifugation and washing.
000, 0.5 g), then centrifuging the mixture to remove unbound polymer and dispersing the solid into a mixture of ethanol, deioned water (5 mL) and glucose (1 g). The mixture was then sealed into a Teflon-lined autoclave. After reaction at 180 °C for 12 h, the carbon coated particles were centrifuged, washed and dispersed in a PVP solution for additional surface modified. Then the suspension was mixed with ethanol (36 mL), water (0.86 mL), aqueous ammonia (0.64 mL) and TEOS (0.86 mL), followed by stirring for 6 h, the SiO2/Au/TiO2/C/SiO2 composite particles were collected by centrifugation and washing.
Calcination and etching
The calcination and etching process was according to the method described in ref. 30. Briefly, the calcination was operated in a muffle furnace at desired temperature for 4 h. The remove of the silica layer was by the chemical etching using NaOH as the etching agent, and the product was labeled as “Auencap/TiO2(C)”.Characterization
Transmission electron microscopy (TEM) experiments were conducted on a JEM-1230 microscope operated at 100 kV. The samples for the TEM measurements were suspended in ethanol and supported onto a Cu grid. X-ray diffraction (XRD) patterns were recorded on a Bruker D8 Advance Diffractometer (Germany) with Cu Kα radiation (λ = 1.5406 Å). The nitrogen adsorption and desorption isotherms were measured at −196 °C on an ASAP 2020 (Micromeritics USA). Prior to measurements, the sample was degassed at 120 °C for 10 h. The specific surface area was determined from the linear part of the BET equation (P/P0 = 0.05–0.25). The pore size distribution was derived from the desorption branch of the N2 isotherm using the Barrett–Joyner–Halenda (BJH) method. UV-Vis absorption spectra analysis was performed on a Shimadzu UV 3600 spectrometer.Catalytic reduction of p-nitrophenol (4-NP)
To study catalytic properties of the prepared Au/TiO2 hollow nanoparticles, reduction of 4-NP by NaBH4 was chosen as a model reaction. In a typical experiment, aqueous solutions of 4-NP (0.2 mL, 0.005 M) and NaBH4 (2 mL, 0.2 M) were added to a quartz cuvette. After adding 0.5 mL of aqueous dispersion of the catalyst particles (0.5 mg mL−1), the yellow solution was gradually faded as the reaction proceeded. The reaction temperature was maintained at 25 °C and UV-Vis spectra of the solution were recorded every 2 min to monitor the progress of the reduction reaction.3. Result and discussion
The work is to synthesize a catalytic system with enhanced thermal stability and high Au loading in which several sub-10 nm Au nanoparticles are encapsulated in each well-crystallized mesoporous TiO2 hollow shell. To obtain the nanocatalyst, multi-steps were carried out, of which the first step was to prepare SiO2/Au/TiO2 composite spheres by sequential deposition of Au and TiO2 on SiO2 bead. Fig. 2a and b showed the typical TEM images obtained after Au and TiO2 deposition, respectively. Tens of Au NPs were seen on each SiO2 microspheres surface. All the Au NPs possessed approximately the same size with about 4.6 nm in size. The Au/SiO2 composite spheres were then coated with a layer of amorphous TiO2 by hydrolyzing TBOT in the presence of HPC. HPC served as a surfactant that modified the surface characteristic of SiO2 and facilitated the deposition of TiO2.33As revealed by TEM images, the deposited TiO2 layer was uniform with about 12 nm thick. No major changes in the size of Au NPs (average size: 5.57 nm) were observed after being coated with TiO2. It was usually suggested that the strong chemical affinity between Au and primary amines of APTES can protected Au NPs from sintering in the SiO2 coating process at room temperature.21,22 Similarly, in our coating process, the strong electrostatic interaction was still robust to prevent the Au NPs from growing even in a relatively high temperature (85 °C). | ||
| Fig. 2 Representative TEM images of (a) Au/SiO2, (b) TiO2/Au/SiO2 and (c) Au/TiO2-P25. | ||
In the second step, the TiO2/Au/SiO2 particles were subjected to a solvothermal process, where the glucose was carbonized to form carbon coated particles. In our previous work, to avoid the dissolution of SiO2 core in the carbonization process, pure ethanol was used as the solvent instead of conventional water, and the thickness of carbon layer can be conveniently controlled by tuning the concentration of glucose.30 In this process, the carbonized of previously dissolved glucose in ethanol may offer the water for the dissolution of residual glucose and promote the further carbonization of glucose. In the work, to accelerate the carbonization rate, we added a small amount of water in the solvothermal process to dissolve more glucose. The result showed the time of carbonization can be reduced to 12 h, and meantime SiO2 core was well reserved (date not show.)
The carbon coated particles were followed by an encapsulation process with the outer SiO2 layer as described by Yin.29,31 In order to crystallize the TiO2 shell, the composite microspheres were subjected to calcination treatment at different temperature. After removal of the SiO2 species in NaOH solution, the crystal structure of the Auencap/TiO2 samples were characterized by XRD measurements. Fig. 3 illustrated the XRD patterns of the final samples at different calcination temperature. The Auencap/TiO2 (C0, 500) sample (direct calcination at 500 °C without the C and SiO2 coating) exhibited a significant improvement in crystallinity of anatase TiO2 even at a lower calcination temperature. The introduction of C and SiO2 layer may offer the space for TiO2 to grow into anatase TiO2, meanwhile inhibit the over-growth of TiO2, as evidenced by the broader XRD peaks corresponding to anatase TiO2. With the calcination temperature increased, the peaks were enhanced correspondingly, demonstrating that the grain size of anatase TiO2 nanocrystals could be well-controlled by tuning the calcination temperature. The XRD pattern also told us that the growth of Au NPs were suppressed for all the TiO2 coated samples, as the XRD peaks corresponding to Au were indistinct. The sandwich structures with the Au NPs embedded inside the TiO2 shell protected the former from moving together and coagulating in the calcination process,32 even for the Auencap/TiO2 (C0, 500) sample, in which the TiO2 shell was extensive crystallization and grain growth.
 | ||
| Fig. 3 XRD patterns of the Auencap/TiO2 samples. (a) Auencap/TiO2 (C0, 500), (b) Auencap/TiO2 (C, 500), (c) Auencap/TiO2 (C, 600) and (d) Auencap/TiO2 (C, 650). | ||
Characterization of TEM also confirmed the minor changes of Au NPs in the preparation process of Auencap/TiO2 catalyst. As shown in Fig. 4, it can be clear that the Auencap/TiO2 samples remained virtually unchanged upon calcinations and etching, demonstrating that the TiO2 shell was quite effective at preserving the size and distribution of the Au NPs even in the growth of TiO2 grains, which should be attributed to the strong interaction between Au and TiO2 by the encapsulation. To better identify Au and TiO2 particles, the magnified TEM image was given in the inset of Fig. 4a. The crystal lattices with d-space equivalent to 0.35 nm and 0.23 nm were corresponded to (101) planes of anatase TiO2 and (110) planes of Au NPs, respectively. Notably, Au NPs of the Auencap/TiO2 (C0, 500) was slightly bigger with respect to the carbon and silica coated samples, accompanied with the severe structure collapse. It was known that significant increase in the grain size of the anatase nanocrystals was inevitable without the protection of the outer carbon and silica layer, which may reduce the interaction between Au and TiO2 particles and result in a growth of Au NPs. Another unfavorable effect was that the significant increase in the grain size of the TiO2 shell may block the outside molecules to reach the buried active Au NPs (see later.)
 | ||
| Fig. 4 TEM images of the prepared Auencap/TiO2 catalysts. (a) Directly calcined TiO2/Au/SiO2 at 500 °C and the magnified TEM image (inset). (b) Calcined the SiO2/TiO2/Au/SiO2 particles at 500 °C. (c) and (d) Calcined the SiO2/TiO2/Au/SiO2 particles at 650 °C. All the samples were calcined in air for 4 h and etched by NaOH in the final step. | ||
N2 physisorption measurements were further used to investigate the structure properties of the Auencap/TiO2 samples. The N2 adsorption/desorption isotherms together with the corresponding pore size distribution of the samples were shown in Fig. 5. As can be observed, both the Auencap/TiO2 (C, 500) and Auencap/TiO2 (C, 650) samples exhibited type IV curves according to the IUPAC nomenclature with two hysteresis loops in the relative pressure range of 0.4–1.0, revealing bimodal pore-size distributions in the mesoporous and macroporous region.34 The first hysteresis loop at a low relative pressure (0.4–0.8), corresponding to the filling of the mesopores formed by the stacking of small TiO2 grains. The second hysteresis loop at a high relative pressure (0.8–1.0), corresponding to the filling of the macropores produced by remove of inner silica core. The surface area are calculated to be as high as about 341.7 m2 g−1 and 297.8 m2 g−1 for the Auencap/TiO2 (C, 500) and Auencap/TiO2 (C, 650) samples, respectively. The slightly decrease of the special surface area should be account for the controlled grain growth of TiO2 by coating with carbon and silica. The detailed pore size distribution calculated based on the BJH method also revealed the growth of TiO2 grains, as evidenced by an increase in the pore diameter after increased the calcination temperature form 500 °C to 650 °C. By contrast, the capillary condensation steps of the Auencap/TiO2 (C0, 500) sample was shifted to a higher relative pressure (P/P0 = 0.6–1.0), which indicated a larger average pore size. Indeed, the pore size distribution curve confirmed that the Auencap/TiO2 (C0, 500) sample possessed mesopores of about 20 nm. The surface area was decreased to 153.2 m2 g−1 correspondingly. As mentioned before, without the protection of carbon and silica shells, the grain size of the anatase nanocrystals were significant increased, and thus lead to severe structure breakage of TiO2 shell and significant decrease in surface area.
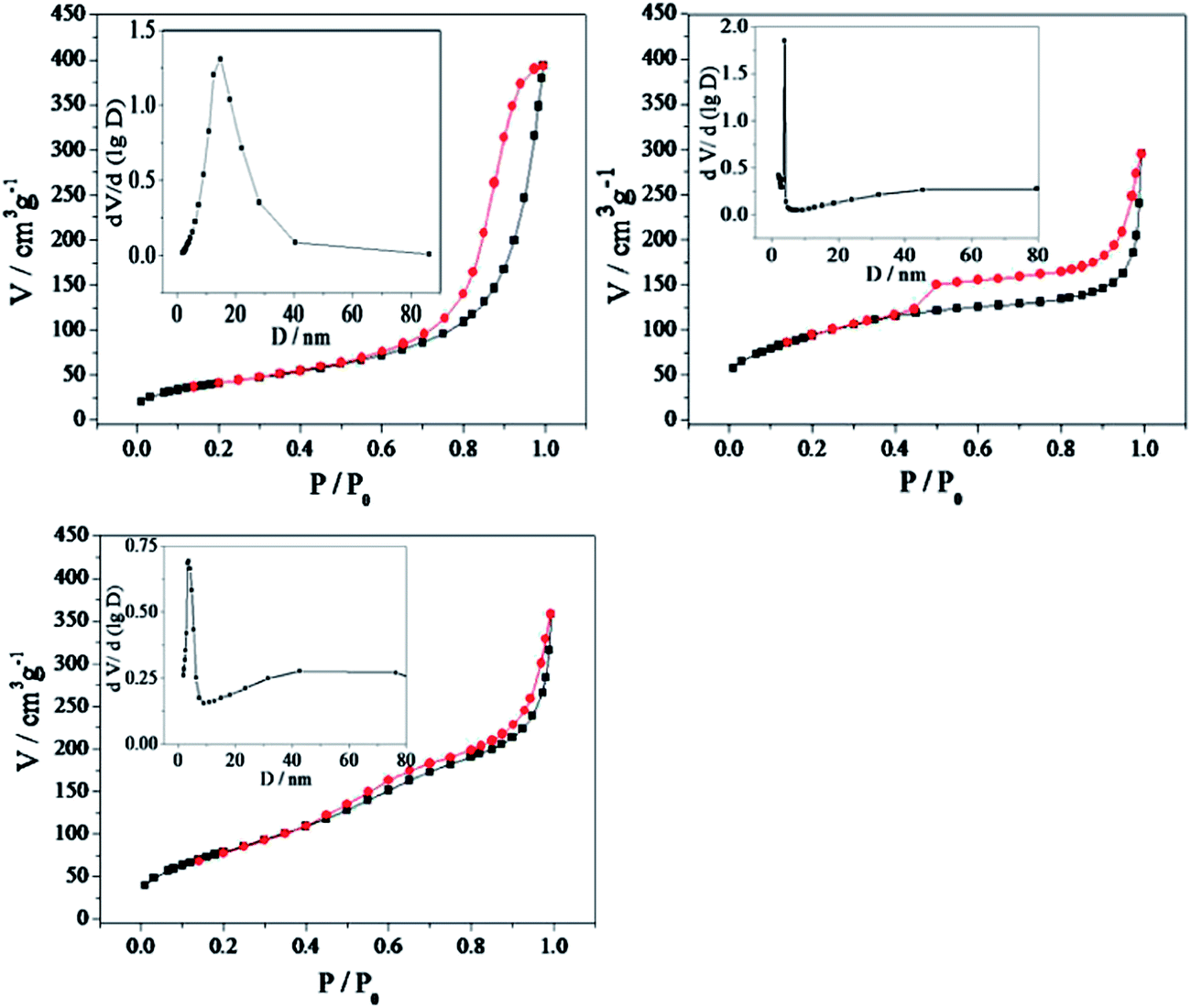 | ||
| Fig. 5 N2 adsorption–desorption isotherms and corresponding pore size distribution curves (inset). (a) Auencap/TiO2 (C0, 500), (b) Auencap/TiO2 (C, 500), (c) Auencap/TiO2 (C, 650). | ||
A better identifying the environment around Au NPs was obtained through the UV-visible absorption characterization of the Auencap/TiO2 samples (Fig. 6). As shown in Fig. 6a, the absorption band at 523 nm was arised from the excitation of surface plasmon mode of the Au NPs in the SiO2 surface. By contrast, when the original Au/SiO2 particles were covered with the TiO2 shell, the surface plasmon peak became too weak to be observed (Fig. 6b), possibly because the dense TiO2 shell confined the transfer of free electrons to the surroundings, which was suggested to give rise to the plasmon resonance.35 However, after converting the TiO2 shell to a porous one, the characteristic absorption band of Au NPs returned (Fig. 6c and d), which suggested that Au NPs became exposed again after calcination treatment. Additionally, the maximum absorption peak was strengthened for the Auencap/TiO2 (C, 650) sample with respect to the Auencap/TiO2 (C0, 500), further demonstrating that Au NPs of the Auencap/TiO2 samples prepared by the protected calcination were more convenient to contact with the environment.
 | ||
| Fig. 6 UV-vis absorption spectra of (a) the original Au/SiO2 particles, (b) TiO2/Au/SiO2 particles, (c) Auencap/TiO2 (C0, 500) particles and (d) Auencap/TiO2 (C, 650) particles. All the particles were dispersed in ethanol solution. | ||
It was also noteworthy that the peak plasmon absorption of the Auencap/TiO2 samples obviously red shifted as compared with the as-prepared Au/SiO2 sample, and the Auencap/TiO2 (C0, 500) sample exhibited a maximum shift. Generally, two reasons, encapsulation and particle aggregation, were accounted for the red shift.36 In this work, the shift in plasmon from 523 nm for the original Au/SiO2 particles to 547 nm for the Auencap/TiO2 (C, 650) was principally upon encapsulation, due to the increase in the refractive index of the composite spheres upon the incorporated Au NPs by crystalline TiO2 shells. In contrast, the further red-shift of the Auencap/TiO2 (C0, 500) sample was mainly caused by the increase in the Au particle dimension. As mentioned before, the Au NPs of the Auencap/TiO2 (C0, 500) sample was slightly bigger than Auencap/TiO2 (C, 650), and thus leading to a red shift of about 10 nm.
The reduction of 4-NP by sodium borohydride was chosen as a model reaction21,27,37 for studying the catalytic performances of the Au/TiO2 catalysts. Since only a small amount of catalyst particles were used, light scattering due to the Au and TiO2 was negligible in comparison to characteristic absorption by 4-NP at 400 nm. No change in the absorption peak at 400 nm was determined after standing for 2 h, suggesting that the reduction reaction did not proceed in the absence of Au catalyst. After addition of a trace amount of catalysts into the solution, the reaction proceeded, as evidenced by the decrease in the absorption at 400 nm and the gradual increase in the absorption at 295 nm. Considering the concentration of NaBH4 largely exceeds that of 4-NP, the reaction rate can be assumed to be independent of NaBH4 concentration. So a pseudo-first-order rate kinetics with respect to 4-NP can be applied to evaluate the catalytic rate.38,39 Fig. 7a showed the linear relationships between ln(Ct/C0) and reaction time in the reaction catalyzed by different Au/TiO2 samples, and the plots well matched the first-order reaction kinetics. According to the linear relationship, we calculated the reaction rate constant k from the slope of the straight lines. The rate constant k of the as-prepared TiO2/Au/SiO2 particles (without calcination treatment) was 0.18 min−1. While for the catalysts after the protecting calcination at 500 °C, the value of k increased to 0.41 min−1, revealing a good activity of Au catalysts after calcination process. Increased the calcination temperature, k further increased to 0.65 min−1 and 1.03 min−1 for 600 °C and 650 °C, respectively, clearly demonstrating that the efficiency of catalysts was improved with the increase of the calcination temperature.
 | ||
| Fig. 7 (a) Relationship of ln(Ct/C0) and reaction time for the reduction of 4-NP catalyzed by different samples. (b) Conversion of 4-NP in 4 successive cycles of reduction with the Auencap/TiO2 (C, 650) and Auencap/TiO2 (C0, 500). | ||
As discussed above, the calcination treatment can help to increase the catalytic activity, which might be ascribed to the following reasons. First, mesopores in TiO2 shells formed by the calcination treatment accelerated the mass transfer rate of the 4-NP. It is known that the pore of the pristine sol–gel derived TiO2 shell (in TiO2/Au/SiO2 catalyst) is restricted to micropore, which would make it difficult for 4-NP to reach the buried Au NPs and result in a relatively lower reactive activity. The calcination process can successfully transform the TiO2 shell into the mesoporous one, and thus better mass diffusion is achieved for the catalysis. Second, the encapsulation of Au NPs with TiO2 layer is quite effective at preventing aggregation of the Au NPs even in the growth of TiO2 grains. The preserving of the size of the Au NPs may retain the number density of active sites for the reduction reaction. Third, the higher crystallinity of TiO2 may promote the electron transfer between Au and TiO2, which may enhance the metal–support interaction. Thus, the calcination treatment has a significantly influence on the catalytic activity and higher catalytic activity can be obtained as for the Auencap/TiO2 (C, 650) catalyst. As a comparison, the amorphous TiO2 in TiO2/Au/SiO2 catalyst not only prevents the diffusion of 4-NP to the buried Au surface, but also possesses the weaker metal–support interaction, thus the as-prepared TiO2/Au/SiO2 exhibited the lowest activity for the reduction of 4-NP.
To investigate the reusability, the catalysts were separated from solution by centrifugation, rinsed with deionized water and reused for the next cycle of catalysis. As shown in Fig. 7b, only a slight decrease in the catalytic activity was observed for Auencap/TiO2 (C, 650) after 4 successive cycles, demonstrating the remarkable stability of Auencap/TiO2 (C, 650). It should be pointed out that though the initial activity of the Auencap/TiO2 (C0, 500) was higher than Auencap/TiO2 (C, 500), the conversion dropped severely after 4 cycles. Such decrease in activity can be explained by the serious loss of catalyst during centrifugation and washing. The Auencap/TiO2 (C0, 500) is composed by the small particles fragments, which are difficult to settle down from the solution with respect to the intact Auencap/TiO2 particles and unavoidably leading to a serious loss of catalyst. Apparently, the unbroken anatase TiO2 shell synthesized by protected-calcination is sufficient for stabilizing Au NPs by preventing their aggregation and erosion.
Finally, to investigate the thermal stability of the catalyst, the Auencap/TiO2 particles were further calcined at 500 °C for 4 h. For comparison, a supported catalyst was prepared by mixing the commercial P25 TiO2 with as-synthesized Au colloids and evaporating the aqueous solution at 70 °C (Au/TiO2-P25). The size of Au NPs in the as-prepared Au/TiO2-P25 catalyst was about 6.3 nm (Fig. 2c), which was comparable to that of TiO2/Au/SiO2 particles (5.57 nm). After calcined, Au NPs already started to coalesce and sintered to 43 nm on the Au/TiO2-P25 sample (Fig. 8a) On the other hand, exposure of the TiO2 protected samples to similar treatments resulted in virtually no changes in the Au diameter after calcination. Over these catalysts, the integrity of the TiO2 shells were varied. Re-calcination of the Auencap/TiO2 (C, 500) samples led to a change in the texture of the hollow structure with respect to the Auencap/TiO2 (C, 650) samples, apparently because the re-growth of TiO2 nanocrystals. The anatase TiO2 was well developed after the initial calcination at 650 °C. As a result, the TiO2 crystal growth in Auencap/TiO2 (C, 650) catalyst were limited. However, the growth of TiO2 in Auencap/TiO2 (C, 500) sample were inhibited in the initial calcination at 500 °C by the SiO2 shell, thus the further TiO2 crystallization was inevitable after removed the SiO2 layer. But it should be underlined that only a small fraction of the hollow structure was affected, most of the TiO2 shells maintained their structure after re-calcination. Thus, the results clearly indicated that our catalysts exhibited excellent sinter resistance and superior thermal stability.
 | ||
| Fig. 8 TEM images of the samples after further calcination at 500 °C. (a) Au/TiO2-P25, (b) Auencap/TiO2 (C, 500), (c) Auencap/TiO2 (C, 650). | ||
4. Conclusions
We have demonstrated a successful synthesis of Auencap/TiO2 catalyst constructed by the sub-10 nm Au NPs imbedded TiO2 nanocrystals via a protected calcination process. The calcination treatment allows the crystallization of TiO2 in a controllable manner, and leads to a well-developed crystallinity, favorable porous structure and small size of the Au NPs. Moreover, the porosity and crystallinity can be well controlled by the change of calcination temperature. The Auencap/TiO2 catalyst shows high activity for the reduction of 4-NP attributed to the small gold size, convenient mass pass and higher crystallinity of TiO2. The anatase TiO2 shell also prevents the Au NPs from sintering after an additional calcination at 500 °C. The combination of anatase TiO2 and Au NPs in our catalyst may extend the application of the particles to photocatalysis.Acknowledgements
The authors are grateful to the financial supports of National Natural Science Foundation of China (Grant no. 21376051, 21306023, 21106017, and 51077013), Natural Science Foundation of Jiangsu (Grant no. BK20131288), Fund Project for Transformation of Scientific and Technological Achievements of Jiangsu Province of China (Grant no. BA2011086), Specialized Research Fund for the Doctoral Program of Higher Education of China (Grant no. 20100092120047), Key Program for the Scientific Research Guiding Found of Basic Scientific Research Operation Expenditure of Southeast University (Grant no. 3207043101) and Instrumental Analysis Fund of Southeast University.References
- M. A. Van Hove, Catal. Today, 2006, 113, 133–140 CrossRef CAS PubMed.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- A. M. Henning, J. Watt, P. J. Miedziak, S. Cheong, M. Santonastaso, M. H. Song, Y. Takeda, A. I. Kirkland, S. H. Taylor and R. D. Tilley, Angew. Chem., Int. Ed., 2013, 52, 1477–1480 CrossRef CAS PubMed.
- M. Abdulla-Al-Mamun, Y. Kusumoto, T. Zannat and M. S. Islam, Phys. Chem. Chem. Phys., 2011, 13, 21026–21034 RSC.
- S. J. Guo and E. K. Wang, Nano Today, 2011, 6, 240–264 CrossRef CAS PubMed.
- J. Qi, J. Chen, G. D. Li, S. X. Li, Y. Gao and Z. Y. Tang, Energy Environ. Sci., 2012, 5, 8937–8941 CAS.
- T. C. Damato, C. C. S. de Oliveira, R. A. Ando and P. H. C. Camargo, Langmuir, 2013, 29, 1642–1649 CrossRef CAS PubMed.
- K. Jalama, N. J. Coville, D. Hildebrandt, D. Glasser, L. L. Jewell, J. A. Anderson, S. Taylor, D. Enache and G. J. Hutchings, Top. Catal., 2007, 44, 129–136 CrossRef CAS PubMed.
- A. Furstner, Chem. Soc. Rev., 2009, 38, 3208–3221 RSC.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS PubMed.
- M. R. Hormozi-Nezhad, P. Karami and H. Robatjazi, RSC Adv., 2013, 3, 7726–7732 RSC.
- X. Y. Liu, P. J. Guo, B. Wang, Z. Jiang, Y. Pei, K. N. Fan and M. H. Qiao, J. Catal., 2013, 300, 152–162 CrossRef CAS PubMed.
- M. D. Xiao, C. M. Zhao, H. J. Chen, B. C. Yang and J. F. Wang, Adv. Funct. Mater., 2012, 22, 4526–4532 CrossRef CAS.
- J. Han, L. Wang and R. Guo, J. Mater. Chem., 2012, 22, 5932–5935 RSC.
- Q. Zhang, I. Lee, J. P. Ge, F. Zaera and Y. D. Yin, Adv. Funct. Mater., 2010, 20, 2201–2214 CrossRef CAS.
- X. L. Fang, Z. H. Liu, M. F. Hsieh, M. Chen, P. X. Liu, C. Chen and N. F. Zheng, ACS Nano, 2012, 6, 4434–4444 CrossRef CAS PubMed.
- C. Chen, X. L. Fang, B. H. Wu, L. J. Huang and N. F. Zheng, ChemCatChem, 2012, 4, 1578–1586 CrossRef CAS.
- I. Lee, M. A. Albiter, Q. Zhang, J. P. Ge, Y. D. Yin and F. Zaera, Phys. Chem. Chem. Phys., 2011, 13, 2449–2456 RSC.
- S. Peng, Y. M. Lee, C. Wang, H. F. Yin, S. Dai and S. H. Sun, Nano Res., 2008, 1, 229–234 CrossRef CAS PubMed.
- I. Lee, J. B. Joo, Y. D. Yin and F. Zaera, Angew. Chem., Int. Ed., 2011, 50, 10208–10211 CrossRef CAS PubMed.
- Y. H. Deng, Y. Cai, Z. K. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Y. Zhao, J. Am. Chem. Soc., 2010, 132, 8466–8473 CrossRef CAS PubMed.
- J. P. Ge, Q. Zhang, T. R. Zhang and Y. D. Yin, Angew. Chem., Int. Ed., 2008, 47, 8924–8928 CrossRef CAS PubMed.
- G. Budroni and A. Corma, Angew. Chem., Int. Ed., 2006, 45, 3328–3331 CrossRef CAS PubMed.
- M. Okumura, S. Tsubota and M. Haruta, J. Mol. Catal. A: Chem., 2003, 199, 73–84 CrossRef CAS.
- M. M. Schubert, S. Hackenberg, A. C. van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113–122 CrossRef CAS.
- K. Qian, L. F. Luo, H. Z. Bao, Q. Hua, Z. Q. Jiang and W. X. Huang, Catal. Sci. Technol., 2013, 3, 679–687 CAS.
- Q. Zhang, T. R. Zhang, J. P. Ge and Y. D. Yin, Nano Lett., 2008, 8, 2867–2871 CrossRef CAS PubMed.
- S. H. Joo, J. Y. Park, C. K. Tsung, Y. Yamada, P. D. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
- J. B. Joo, Q. Zhang, I. Lee, M. Dahl, F. Zaera and Y. D. Yin, Adv. Funct. Mater., 2012, 22, 166–174 CrossRef CAS.
- Z. W. Zhang, Y. M. Zhou, Y. W. Zhang, S. J. Zhou, J. J. Shi, J. Kong and S. C. Zhang, Dalton Trans., 2013, 42, 5004–5012 RSC.
- J. B. Joo, Q. Zhang, M. Dahl, I. Lee, J. Goebl, F. Zaera and Y. D. Yin, Energy Environ. Sci., 2012, 5, 6321–6327 CAS.
- Q. Zhang, D. Q. Lima, I. Lee, F. Zaera, M. F. Chi and Y. D. Yin, Angew. Chem., Int. Ed., 2011, 50, 7088–7092 CrossRef CAS PubMed.
- J. P. Cheng, R. Ma, M. Li, J. S. Wu, F. Liu and X. B. Zhang, Chem. Eng. J., 2012, 210, 80–86 CrossRef CAS PubMed.
- J. D. Zhuang, Q. F. Tian, H. Zhou, Q. Liu, P. Liu and H. M. Zhong, J. Mater. Chem., 2012, 22, 7036–7042 RSC.
- R. T. Tom, A. S. Nair, N. Singh, M. Aslam, C. L. Nagendra, R. Philip, K. Vijayamohanan and T. Pradeep, Langmuir, 2003, 19, 3439–3445 CrossRef CAS.
- K. S. Mayya, D. I. Gittins and F. Caruso, Chem. Mater., 2001, 13, 3833–3836 CrossRef CAS.
- N. Pradhan, A. Pal and T. Pal, Langmuir, 2001, 17, 1800–1802 CrossRef CAS.
- T. Y. Yu, J. Zeng, B. Lim and Y. N. Xia, Adv. Mater., 2010, 22, 5188–5192 CrossRef CAS PubMed.
- J. Zeng, Q. Zhang, J. Y. Chen and Y. N. Xia, Nano Lett., 2010, 10, 30–35 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |