Synthesis of the trisaccharide moiety and a cholesteryl analog of phyteumosides†
Sanjoy Adak,
Madhu Emmadi and
Suvarn S. Kulkarni*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai, 400076, India. E-mail: suvarn@chem.iitb.ac.in; Fax: +91-22-2576-7152; Tel: +91-22-2576-7166
First published on 8th January 2014
Abstract
A first synthesis of the trisaccharide moiety of the phyteumosides and its cholesteryl analog is described using easily accessible and regioselectively protected D-glucose, L-rhamnose and D-galactose building blocks via a linear glycosylation approach. The two glycosyl donors were prepared as thiophenyl (SPh) glycosides. Trichloroacetimidate (TCAI) coupling was employed for the first glycosylation step while thioglycoside activation was used for the second glycosylation to assemble the O-allyl trisaccharide. After removal of the anomeric allyl group, the trisaccharide was converted into a TCAI donor. Using cholesterol as an acceptor in combination with acetonitrile as a participating solvent to achieve β-selectivity in glycosylation, a phyteumoside analog was obtained.
Introduction
Saponins (glycosides of steroids and terpenes) play key roles as secondary metabolites in plants and marine organisms.1 In 2011, Potterat and co-workers isolated and characterized two novel triterpene glycosides phyteumosides A (1a) and B (1b) from Phyteuma orbiculare (Fig. 1).2 Phyteumosides A and B comprise a trisaccharide moiety glycosidically β-O-linked to the unique triterpene aglycons. This trisaccharide is also a part of the tetrasaccharide glycone of macrophyllicin, a saponin isolated from Primula macrophylla in 1993.2b No synthetic studies on the trisaccharide moiety or 1a and 1b are reported till date. Herein, we report a first synthesis of the trisaccharide moiety and a cholesteryl analog 1c of the phyteumosides. | ||
| Fig. 1 Structure of the triterpenoid saponins from Phyteuma orbiculare. | ||
Results and discussions
We envisaged that the saponin analog 1c (Scheme 1) could be assembled by glycosylation of TCAI analog of trisaccharide 2 with cholesterol 3 using solvent effect to ensure the β-selectivity in lieu of a 2-O- participating group. O-Allyl trisaccharide 2 could in turn be synthesized by stereoselective coupling of appropriately protected monosaccharide building blocks 4, 5, and 6, from the reducing end to non-reducing end. | ||
| Scheme 1 Retrosynthesis of phyteumoside analog 1c. | ||
The building blocks 4, 5 and 6 were synthesized following reported procedures. Thus, compound 4 was obtained from L-rhamnose by a one-pot per-O-acetylation followed by nulceophilic displacement of anomeric acetate with thiophenol using Cu(OTf)2 (ref. 3) whereas compound 5 (ref. 4) was synthesized from similarly obtained D-glucosyl thioglycoside by 4,6-O-benzylidenation,5 regioselective silylation at O3,6 and followed by acetylation at O2. Compound 6 was accessed from the corresponding O-allyl galactoside7 via regioselective 3,4-O-isopropylidenation8 and O6 silylation in good yields.
Coupling of donor 5 with acceptor 6 to synthesize disaccharide 7 was performed next (Scheme 2). However, to our dismay, no coupling product could be obtained through thioglycoside activation under various conditions (NIS/TfOH, NIS/TMSOTf, conversion to glycosyl bromide and activation with AgOTf). So, the thioglycoside 5 was converted to trichloroacetimidate,9 which upon activation with TMSOTf in the presence of acceptor 6 delivered the desired β-linked disaccharide 7 in reasonable yields (51% from 5, over 3 steps).
 | ||
| Scheme 2 Synthesis of disaccharide acceptor 8a. | ||
After the successful synthesis of disaccharide 7, the 2′-O-acetate was removed using NaOMe in MeOH at RT to afford the desired disaccharide acceptor 8a in 55% yield, along with a side product 8b (40%) resulting from the concomitant migration of TBDMS group from O3′ to O2′ under the prevailing basic conditions. Although not very common, such silyl group migrations have been well documented in literature.10,11 We propose that this O3′ to O2′ silyl migration probably proceeds in a concerted manner through an intramolecular attack of the O2′ alkoxide II on O3′-siliyl group to give a transient five membered silyl intermediate III which subsequently opens on the other side to give alkoxide IV (Scheme 3).10,11e Since alkoxides II and IV have comparable thermodynamic stability, these species remain in equilibrium with each other and one obtains a mixture of 8a and 8b. Compounds 8a and 8b were easily separated by column chromatography and their structures were unambiguously confirmed by analyzing their 2D NMR spectra. The 1H–1H COSY spectrum of 8b clearly showed correlation of the OH proton with the C3′-H (ESI†).
All attempts to suppress the formation of the migration by-product 8b were unsuccessful. In order to avoid such migration, we also tried the reaction on the disaccharide having OBz at 2′ position. Despite applying several deacylation reaction conditions (NaOMe in MeOH at rt, NaOMe in MeOH under reflux condition, aqueous NaOH solution) we were not able to cleave the OBz group. Although, not useful from the point of view of present study, compound 8b is nevertheless an advanced building block which could be advantageously utilized as a glycosyl acceptor in the synthesis of related complex carbohydrates (Scheme 3).
 | ||
| Scheme 3 Proposed mechanism of silyl group migration. | ||
With the desired acceptor 8a in hand we went ahead to synthesize the trisaccharide moiety of phyteumoside. Acceptor 8a was glycosylated with thioglycoside donor 4 using NIS and TMSOTf promoter12 system to afford the trisaccharide 2 in 53% yield (Scheme 4). The modest yield could be attributed to simultaneous partial activation of the allyl double bond in 8a or 2 by NIS. For the global deprotection, trisaccharide 2 was first treated with TBAF,13 followed by 80% AcOH14 to sequentially remove TBDMS and acetal groups, respectively. The so obtained polyol was treated with acetic anhydride and pyridine to obtain the per-O-acetylated trisaccharide 9 in 78% yield over 3 steps.
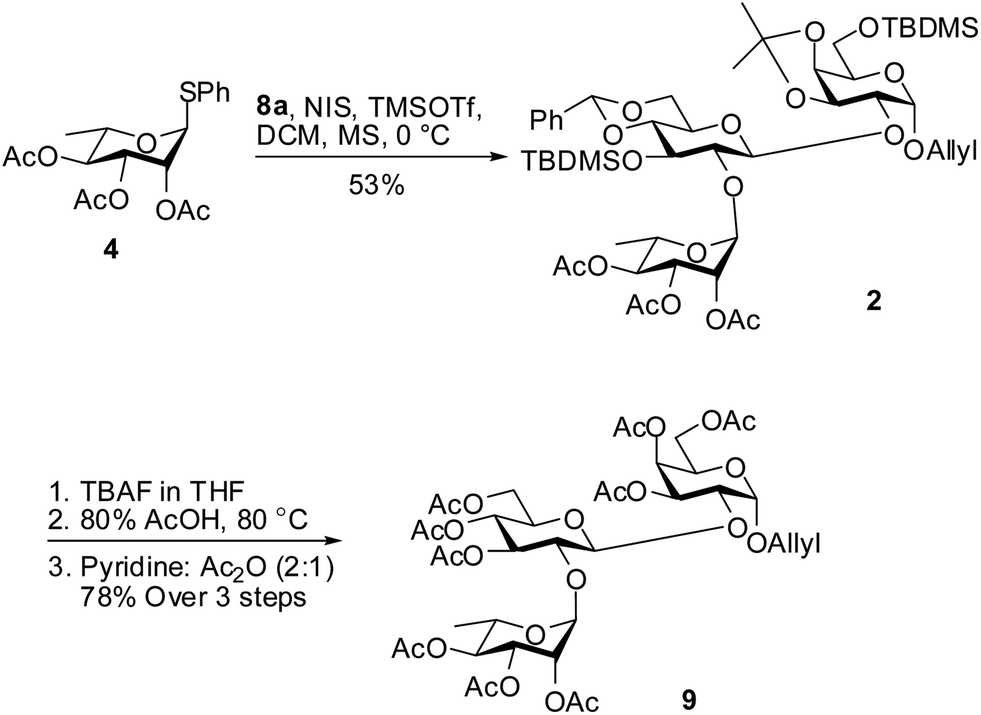 | ||
| Scheme 4 Synthesis of trisaccharide with the linker 9. | ||
For the preparation of phyteumoside analog 1c (Scheme 5), the allyl group in trisaccharide 2 was oxidatively removed by treatment with OsO4, NMO and NaIO4 (ref. 15) to afford hemiacetal 10 (α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β = 0.6:1) in 79% yield. Compound 10 was converted to the corresponding imidate by treating with trichloroacetonitrile and the so formed imidate was coupled with cholesterol 3, in acetonitrile as participating solvent,16 to afford exclusively the β-linked phyteumoside analog 11 in 45% yields over 2 steps. Deprotection of TBDMS and acetal groups followed by acetylation under similar conditions afforded the saponin analog 12 as a per-O-acetate derivative in good yields. Treatment of 12 with NaOMe in MeOH at RT afforded 1c in 86% yield.
:β = 0.6:1) in 79% yield. Compound 10 was converted to the corresponding imidate by treating with trichloroacetonitrile and the so formed imidate was coupled with cholesterol 3, in acetonitrile as participating solvent,16 to afford exclusively the β-linked phyteumoside analog 11 in 45% yields over 2 steps. Deprotection of TBDMS and acetal groups followed by acetylation under similar conditions afforded the saponin analog 12 as a per-O-acetate derivative in good yields. Treatment of 12 with NaOMe in MeOH at RT afforded 1c in 86% yield.
 | ||
| Scheme 5 Synthesis of saponin analog 12. | ||
All the new compounds were thoroughly characterized using spectral means. A typical NMR characterization sequence involved 1H, 13C, HMQC and 1H–1H COSY analysis to unambiguously assign all the sugar protons (see ESI†).
Conclusions
In summary, we have synthesized the trisaccharide moiety and a cholesteryl analog of phyteumosides for the first time, by using easily accessible monosaccharide building blocks. The trisaccharide derivative 10 can be coupled with a variety of aglycons to prepare phyteumoside A and B and various other analogs for biological studies.Experimental
All reactions were conducted under a dry nitrogen atmosphere. Solvents (CH2Cl2 >99%, THF 99.5%, acetonitrile 99.8%, DMF 99.5%) were purchased in capped bottles and dried under sodium or CaH2. All other solvents and reagents were used without further purification. All glassware was oven-dried before use. TLC was performed on precoated aluminum plates of silica gel 60 F254 (0.25 mm, E. Merck). Developed TLC plates were visualized under a short-wave UV lamp and by heating plates that were dipped in ammonium molybdate/cerium(IV) sulfate solution. Silica gel column chromatography was performed using silica gel (100–200 mesh) and employed a solvent polarity correlated with TLC mobility. NMR experiments were conducted on 400 and 500 MHz instrument using CDCl3 (D, 99.8%) CD3OD (D, 99.8%) as solvents. Chemical shifts are relative to the deuterated solvent peaks and are in parts per million (ppm). Mass spectra were acquired in the ESI mode using Q-TOF analyzer. Specific rotation experiments were measured at 589 nm (Na) and 25 °C/20 °C. IR spectra were recorded on an FT-IR spectrometer using CsCl plates.Allyl 2-O-acetyl-4,6-O-benzylidene-3-O-tert-butyldimethylsilyl-β-D-glucopyranosyl-(1→2)-6-O-tert-butyldimethylsilyl-3,4-O-isopropylidene-α-D-galactopyranoside (7)
To a cooled solution of 5 (2.37 g, 4.59 mmol) in THF and water (40 mL, 4:1) at 0 °C, was slowly added NBS (2.45 g, 13.77 mmol). After stirring at room temperature for 15 min, the reaction mixture was diluted with EtOAc (80 mL) and washed with aq. NaHCO3. Separated organic layer dried over Na2SO4, filtered and concentrated. The crude product which was obtained after solvents removal was dissolved in CH2Cl2 (43 mL). To this clear solution K2CO3 (1.9 g), and NCCCl3 (2.12 mL) were added and stirred at ambient temperature overnight. The reaction mixture was filtered through celite pad. The filtrate was concentrated in vacuo and the crude product was used for the next reaction. To the residue which was obtained after solvents removal was added acceptor 6 (1.03 g, 2.75 mmol), 4 Å MS and CH2Cl2 (30 mL). The resulted turbid was stirred at room temperature for 30 min and brought to −15 °C. To this cooled solution TMSOTf (58 μL) was added at −15 °C and stirred for 1.5 h at the same temperature. After complete consumption of starting material reaction mixture was quenched by adding Et3N. The reaction mixture was filtered through celite and washed with CH2Cl2. The filtrate was concentrated to give a residue that was purified by silica gel column chromatography (petroleum ether:EtOAc = 8:1) to afford 7 (1.81 g, 51% over three steps from 5) as a white foam: [α]25D 20.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.48–7.41 (m, 2H, ArH), 7.36–7.32 (m, 3H, ArH), 5.93–5.83 (m, 1H, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.51 (s, 1H, benzylidene), 5.30 (dd, J = 17.2, 1.6 Hz, 1H, HCCH2), 5.18 (dd, J = 10.4, 1.6 Hz, 1H, HCCH2), 4.96 (t, J = 8.6 Hz, 1H, H-2′), 4.87 (d, J = 3.4 Hz, 1H, H-1), 4.76 (d, J = 8.6 Hz, 1H, H-1′), 4.28-4.22 (m, 2H, H-3 & H-6a′), 4.19–4.11 (m, 2H, H-6a & H2CCH2), 4.05–3.97 (m, 2H, H-6b & H2CCH2), 3.90–3.83 (m, 2H, H-3′ & H-5), 3.79–3.73 (m, 3H, H-2, H-4, H-6b′), 3.52 (t, J = 8.6, 1H, H-4′), 3.41–3.35 (m, 1H, H-5′), 2.08 (s, 3H, CH3), 1.49 (s, 3H, CH3), 1.31 (s, 3H, CH3), 0.89 (s, 9H, (CH3)3C), 0.81 (s, 9H, (CH3)3C), 0.07 (s, 6H, 2CH3), 0.02 (s, 3H, CH3), −0.02 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 169.6, 137.2, 133.9, 129.1, 128.3, 126.3, 117.5, 109.0, 102.0, 101.9, 97.1, 81.6, 78.1, 76.8, 75.5, 74.6, 73.5, 72.7, 68.8, 68.6, 68.2, 66.4, 62.4, 29.8, 28.5, 26.7, 25.9, 25.7, 21.2, 18.4, 18.1, −4.0, −4.8, −5.1, −5.2. HRMS calcd for C39H64NaO12Si2 [M + Na]+ 803.3829, found 803.3837.
CH2), 5.51 (s, 1H, benzylidene), 5.30 (dd, J = 17.2, 1.6 Hz, 1H, HCCH2), 5.18 (dd, J = 10.4, 1.6 Hz, 1H, HCCH2), 4.96 (t, J = 8.6 Hz, 1H, H-2′), 4.87 (d, J = 3.4 Hz, 1H, H-1), 4.76 (d, J = 8.6 Hz, 1H, H-1′), 4.28-4.22 (m, 2H, H-3 & H-6a′), 4.19–4.11 (m, 2H, H-6a & H2CCH2), 4.05–3.97 (m, 2H, H-6b & H2CCH2), 3.90–3.83 (m, 2H, H-3′ & H-5), 3.79–3.73 (m, 3H, H-2, H-4, H-6b′), 3.52 (t, J = 8.6, 1H, H-4′), 3.41–3.35 (m, 1H, H-5′), 2.08 (s, 3H, CH3), 1.49 (s, 3H, CH3), 1.31 (s, 3H, CH3), 0.89 (s, 9H, (CH3)3C), 0.81 (s, 9H, (CH3)3C), 0.07 (s, 6H, 2CH3), 0.02 (s, 3H, CH3), −0.02 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 169.6, 137.2, 133.9, 129.1, 128.3, 126.3, 117.5, 109.0, 102.0, 101.9, 97.1, 81.6, 78.1, 76.8, 75.5, 74.6, 73.5, 72.7, 68.8, 68.6, 68.2, 66.4, 62.4, 29.8, 28.5, 26.7, 25.9, 25.7, 21.2, 18.4, 18.1, −4.0, −4.8, −5.1, −5.2. HRMS calcd for C39H64NaO12Si2 [M + Na]+ 803.3829, found 803.3837.
Allyl 4,6-O-benzylidene-3-O-tert-butyldimethylsilyl-β-D-glucopyranosyl-(1→2)-6-O-tert-butyldimethylsilyl-3,4-O-isopropylidene-α-D-galactopyranoside (8a)
To a stirred solution of disaccharide 7 (1.5 g, 1.92 mmol) in dry MeOH (9 ml) was added NaOMe (0.24 g) and the mixture was stirred at room temperature for 2 h. After complete consumption of starting material, it was neutralized by addition of Dowex resin. Then the reaction mixture was filtered and concentrated under reduced pressure. The residue was purified by column chromatography (petroleum ether:EtOAc = 6:1) to afford compound 8a as a white foam (0.77 g, 55% yield): [α]25D 20.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.49–7.42 (m, 2H, ArH), 7.37–7.31 (m, 3H, ArH), 5.95–5.85 (m, 1H, HC→CH2), 5.50 (s, 1H, benzylidene), 5.31 (dd, J = 17.2, 1.5 Hz, 1H, HC→CH2), 5.20 (dd, J = 10.3, 1.5 Hz, 1H, HC→CH2), 4.94 (d, J = 3.4 Hz, 1H, H-1), 4.65 (d, J = 7.8 Hz, 1H, H-1′), 4.40 (dd, J = 8.2, 5.2 Hz, 1H, H-6a′), 4.28–4.15 (m, 3H, H-3, H-6a & CH2), 4.06–4.00 (m, 2H, CH2 & H-6b), 3.88–3.65 (m, 5H, H-3′, H-2, H-4, H-5 & H-6b′), 3.55–3.44 (m, 2H, H-2′ & H-4′), 3.40–3.34 (m, 1H, H-5′), 1.51 (s, 3H, CH3), 1.33 (s, 3H, CH3), 0.90 (s, 9H, (CH3)3C), 0.87 (s, 9H, (CH3)3C), 0.10 (s, 3H, CH3), 0.08 (s, 6H, 2CH3), 0.04 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 137.4, 133.8, 129.1, 128.3, 126.3, 118.0, 109.3, 104.2, 101.7, 96.9, 81.5, 77.9, 76.9, 75.5, 75.4, 74.4, 73.6, 68.9, 68.4, 68.3, 66.6, 62.4, 28.5, 26.6, 26.1, 25.9, 18.5, 18.4, −4.2, −4.5, −5.1, −5.3; HRMS calcd for C37H62NaO11Si2 [M + Na]+ 761.3723, found 761.3721.
Allyl 4,6-O-benzylidene-2-O-tert-butyldimethylsilyl-β-D-glucopyranosyl-(1→2)-6-O-tert-butyldimethylsilyl-3,4-O-isopropylidene-α-D-galactopyranoside (8b)
(0.56 g, 40% yield): [α]20D + 11.6 (c 2.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.49–7.47 (m, 2H, ArH), 7.37–7.34 (m, 3H, ArH), 5.94–5.85 (m, 1H, HCCH2), 5.51 (s, 1H, benzylidene), 5.29 (dd, J = 17.2, 1.5 Hz, 1H, HCCH2), 5.23 (dd, J = 10.3, 1.5 Hz, 1H, HC = CH2), 4.39 (dd, J = 8.4, 5.2 Hz, 1H, H-6a), 4.29–4.12 (m, 3H, H-3, H-6a′ & CH2), 4.07–3.93 (m, 4H, H-2, H-5, H-6b & CH2), 3.90–3.71 (m, 3H, H-3′, H-6b′ & H-5), 3.57–3.47(m, 3H, H-2′, H-4′ & H-5′), 2.51 (brs, 1H, 3′–OH) 1.49 (s, 3H, CH3), 1.33 (s, 3H, CH3), 0.93 (s, 9H, (CH3)3C), 0.91 (s, 9H, (CH3)3C), 0.19 (s, 3H, CH3), 0.15 (s, 3H, CH3), 0.09 (s, 6H, 2CH3); 13C NMR (100 MHz, CDCl3) δ 137.2, 134.0, 129.2, 128.4, 126.3, 117.4, 108.9, 102.0, 101.8, 97.5, 80.7, 76.0, 74.8, 74.6, 73.6, 68.9, 68.6, 68.2, 66.1, 62.4, 28.3, 26.7, 26.1, 25.9, 18.4, 18.3, −4.0, −4.6, −5.2, −5.3; HRMS calcd for C37H62NaO11Si2 [M + Na]+ 761.3723, found 761.3729.
Allyl 2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl (1→2)-4,6-O-benzylidene-3-O-tert-butyldimethylsilyl-β-D-glucopyranosyl-(1→2)-6-O-tert-butyldimethylsilyl-3,4-O-isopropylidene-α-D-galactopyranoside (2)
Acceptor 8a (21 mg, 0.028 mmol) and donor 4 (13 mg, 0.035 mmol) were combined and co-evaporated with toluene (3 × 5 ml) and dried under high vacuum for 2 h. The resulting residue was dissolved in anhydrous CH2Cl2 (1.0 mL) and stirred over activated molecular sieves for 30 min. The anhydrous mixture was cooled to 0 °C, NIS (10 mg, 1.23 mmol) and TMSOTf (2 μL) were added and the mixture was stirred at 0 °C for 90 min. After complete consumption of starting material, it was quenched by adding triethylamine. The mixture was filtered and washed with CH2Cl2. The filtrate was concentrated to give a residue that was purified by silica gel column chromatography (petroleum ether:EtOAc = 8:1) to afford 2 as white foam (15 mg, 53%): [α]25D −8.6 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.44–7.40 (m, 2H, ArH), 7.36–7.30 (m, 3H, ArH), 5.91–5.83 (m, 1H, HCCH2), 5.41–5.39 (m, 2H, H-1′′ & benzylidene), 5.32–5.25 (m, 3H, H-2′′, H-3′′ & HCCH2), 5.15 (dd, J = 10.0, 1.3 Hz, 1H, HCCH2), 5.05 (t, J = 10.0 Hz, 1H, H-4′′), 4.93 (d, J = 3.3 Hz, 1H, H-1), 4.74 (d, J = 7.8 Hz, 1H, H-1′), 4.61–4.58 (m, 1H, H-5′′), 4.3–4.12 (m, 4H, H-6a, H-6b′, H-6b & CH2), 4.03–3.96 (m, 3H, H-3, H-3′ & CH2), 3.88–3.66 (m, 5H, H-2′, H-2, H-4, H-5 & H-6a′), 3.43–3.34 (m, 2H, H-4′ & H-5′), 2.10 (s, 3H, CH3), 2.01 (s, 3H, CH3), 1.95 (s, 3H, CH3), 1.54 (s, 3H, CH3), 1.31 (s, 3H, CH3), 1.18 (d, J = 6.2 Hz, 3H, CH3), 0.89 (s, 9H, (CH3)3C), 0.72 (s, 9H, (CH3)3C), 0.07 (s, 6H, 2 CH3), −0.07 (s, 3H, CH3), −0.09 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 170.1, 170.0, 169.8, 137.0, 134.0, 129.4, 128.3, 126.6, 117.5, 109.2, 103.0, 102.6, 98.1, 97.5, 81.4, 78.9, 77.3, 76.4, 74.9, 73.7, 71.2, 69.7, 68.8, 68.4, 68.3, 66.5, 66.0, 62.5, 28.5, 26.5, 26.0, 25.9, 21.0, 20.98, 20.93, 18.4, −3.7, −4.3, −5.2, −5.3; HRMS calcd for C49H78NaO18Si2 [M + Na]+ 1033.4619, found 1033.4619.
Allyl 2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl (1→2)-3,4,6-tri-O-acetyl-β-D-glucopyranosyl-(1→2)-3,4,6-tri-O-acetyl-α-D-galactopyranoside (9)
TBAF (370 μL of 1 M solution in THF) was added to a stirred solution of the trisaccharide 2 (0.14 g, 0.013 mmol) in THF (2.5 mL). The mixture was stirred at rt for 4 h, then evaporated to dryness under reduced pressure. After complete removal of solvents, crude product was dissolved in 80% AcOH (13 mL), and the solution was heated at 80 °C for 3 h. The reaction mixture was then concentrated, and the resulting compound was dissolved in 2:1 pyridine:Ac2O (12 mL). After stirring for 16 h, the reaction mixture was concentrated and the product was purified by silica gel chromatography (40% ethyl acetate in pet ether) to give compound 9 (97 mg, 78% Over 3 steps): [α]25D 26.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.88–5.79 (m, 1H, HCCH2), 5.47 (d, J = 1.5 Hz, 1H, H-4), 5.29 (d, J = 17.2 Hz, 1H, HCCH2), 5.20 (t, J = 10.0 Hz, 1H, H-3′), 5.14 (d, J = 12.0 Hz, 1H, HCCH2), 5.09–5.04 (m, 2H, H-3, H-3′′), 4.99–4.89 (m, 4H, H-1, H-2′′, H-4′ & H-4′′), 4.83 (s, 1H, H-1′′), 4.56 (d, J = 7.6 Hz, 1H, H-1′), 4.22–4.10 (m, 4H, H-5, H-6a′, H-6b′, CH2), 4.07–4.0 (m, 3H, H-6a, H-6b & CH2), 3.96–3.87 (m, 2H, H-2 & H-5′′), 3.70–3.63 (m, 2H, H-2′ & H-5′), 2.11 (s, 3H, CH3), 2.07 (s, 6H, 2CH3), 2.03 (s, 9H, 3 CH3), 2.02 (s, 3H, CH3), 1.98 (s, 3H, CH3), 1.97 (s, 3H, CH3), 1.11 (d, J = 6.0 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 170.6, 170.5, 170.4, 170.3, 170.2, 170.1, 169.8, 169.7, 133.6, 117.3, 101.7, 98.4, 98.1, 76.6, 74.4, 73.2, 71.7, 69.3, 69.0, 68.9, 68.3, 68.2, 66.9, 66.2, 61.8, 61.7, 20.9, 20.87, 20.81, 20.78, 20.74, 20.71, 20.6, 16.8; HRMS calcd for C39H54O24 [M + Na]+ 929.2897, found 929.2886.
2,3,4-Tri-O-acetyl-α-L-rhamnopyranosyl (1→2)-4,6-O-benzylidene-3-O-tert-butyldimethylsilyl-β-D-glucopyranosyl (1→2)-6-O-tert-butyldimethylsilyl-3,4-O-isopropylidene-α/β-D-galactopyranose (10)
To a solution of 2 (0.36 g, 0.35 mmol) in dioxane (4 ml) and water (0.4 ml) was added a mixture of 4-methylmorpholine N-oxide (124 mg, 1.0 mmol) and OsO4 (0.1 mL, 100 mg mL−1 solution in t-BuOH) followed by addition of a suspension of NaIO4 (227 mg, 1.0 mmol) in water (1 mL). The mixture was allowed to stir at 60 °C for 16 h and then diluted with brine and extracted with CH2Cl2. Separated organic layer dried over Na2SO4, filtered and concentrated. The obtained residue was purified by column chromatography on silica gel with petroleum ether–ethyl acetate (4:1) to give the compound 10 as a foam (0.28 g 79%): [α]25D −27.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.43–7.40 (m, 2H, ArH), 7.36–7.26 (m, 3H, ArH), 5.40–5.39 (m, 2H), 5.34–5.28 (m, 2H), 5.08–5.02 (m, 1H), 4.74 (d, J = 7.8 Hz, 1H), 4.54–4.15 (m, 5H), 4.03–3.95 (m, 1H), 3.89–3.64 (m, 6H), 3.46–3.41 (m, 2H), 2.11 (s, 3H, CH3), 2.02 (s, 3H, CH3), 1.95 (s, 3H, CH3), 1.54 (s, 3H, CH3), 1.31 (s, 3H, CH3), 1.18 (d, J = 6.2 Hz, 3H, CH3), 0.89 (s, 9H, (CH3)3C), 0.72 (s, 9H, (CH3)3C), 0.07 (s, 6H, 2CH3), −0.07 (s, 3H, CH3), −0.09 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 170.1, 170.0, 169.8, 137.0, 134.0, 129.4, 128.3, 126.6, 117.5, 109.2, 103.0, 102.6, 98.1, 97.5, 81.4, 78.9, 76.8, 76.4, 74.9, 73.7, 71.2, 69.7, 68.8, 68.4, 68.3, 66.5, 66.0, 62.5, 28.5, 26.5, 26.0, 25.9, 21.0, 20.98, 20.93, 18.4, −3.8, −4.5, −5.3, −5.4; HRMS calcd for C46H74O18Si2 [M + Na]+ 993.4306, found 993.4283.
Cholesteryl 2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl (1→2)-4,6-O-benzylidene-3-O-tert-butyldimethylsilyl-β-D-glucopyranosyl (1→2)-6-O-tert-butyldimethylsilyl-3,4-O-isopropylidene-β-D-galactopyranoside (11)
A suspension of 10 (0.11 g, 0.12 mmol), K2CO3 (50 mg), and NCCCl3 (56 μL) in CH2Cl2 (2 mL) was allowed to stir at ambient temperature overnight. The mixture was filtered through celite and washed with CH2Cl2. Filtrate was concentrate under reduced pressure. The suspension of residue which was obtained after solvent removal, cholesterol (68 mg, 0.175 mmol) and 4 Å molecular sieves in CH2Cl2:CH3CN (2 mL, 1:1) were stirred at room temperature for 30 min. Then, the solution was cooled 0 °C and then TMSOTf (2 μL, 0.012 mmol) was added dropwise. After 30 min, the mixture was diluted with CH2Cl2, filtered through celite and concentrated. The residue was purified by silica gel chromatography (15% ethyl acetate in pet ether) to give the desired product 11 as a foam (60 mg, 45%): [α]25D −5.4 (c 0.12, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.46–7.43 (m, 2H, ArH), 7.36–7.29 (m, 3H, ArH), 5.42 (s, 1H, benzylidene), 5.36–5.28 (m, 4H, H-1′′, H-2′′, H-3′′ & HC), 5.05 (t, J = 10.0 Hz, 1H, H-4′′), 4.99 (d, J = 7.8 Hz, 1H, H-1′), 4.38 (d, J = 7.8 Hz, 1H, H-1), 4.27–4.19 (m, 3H, H-5, H-5′′ & H-4), 3.98 (t, J = 10.0 Hz, 1H, H-3′), 3.86–68 (m, 5H, H-2, H-6a, H-6b, H-6a′ & H-6b′), 3.57–3.52 (m, 4H, H-2′, H-3, H-4′ & H-5′), 2.30–2.28 (m, 2H), 2.12 (s, 3H, CH3), 2.03 (s, 3H, CH3), 1.96 (s, 3H, CH3), 1.87–1.79 (m, 3H), 1.68 (s, 3H), 1.51–1.45 (m, 6H), 1.48 (s, 3H, CH3), 1.33 (s, 3H, CH3), 1.29-1.21 (m, 21H), 1.11 (d, J = 6.0 Hz, 3H, CH3), 0.91–0.83 (m, 16H), 0.75 (s, 9H), 0.67 (s, 3H), 0.06 (s, 6H), −0.04 (s, 3H), −0.07 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.2, 170.0, 169.9, 140.8, 137.2, 129.3, 128.3, 126.6, 121.9, 110.2, 109.1, 102.5, 100.2, 99.3, 97.7, 81.4, 79.4, 79.2, 76.3, 73.6, 73.4, 71.6, 69.7, 69.3, 69.1, 66.4, 66.2, 62.4, 56.9, 56.3, 50.3, 42.5, 39.9, 39.6, 39.0, 37.5, 36.9, 36.3, 35.9, 32.1, 32.0, 29.9, 28.4, 28.2, 27.8, 26.3, 26.0, 25.9, 24.2, 23.9, 23.0, 22.7, 21.2, 21.1, 21.0, 20.9, 19.5, 18.8, 18.4, 18.3, 17.7, 12.0, −3.6, −4.4, −5.1, −5.2, −5.3; HRMS calcd for C73H118O18Si2 [M + Na]+ 1361.7749, found 1361.7788.
Cholesteryl 2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl-(1→2)-3,4,6-tri-O-acetyl-β-D-glucopyranosyl-(1→2)-3,4,6-tri-O-acetyl-β-D-galactopyranoside (12)
TBAF (75 μL of 1 M solution in THF) was added to a stirred solution of 11 (37 mg, 0.027 mmol) in THF (0.5 mL). The mixture was stirred at room temperature for 4 h, then evaporated to dryness under reduced pressure. After that, the crude product was dissolved in 80% AcOH in H2O (3 mL), and the solution was heated at 80 °C for 3 h. Then the reaction mixture was concentrated, and the resulting white solid was dissolved in 2:1 pyridine:Ac2O (3 mL). After stirring for 16 h, the reaction mixture was concentrated and the product was purified by silica gel chromatography (40% ethyl acetate in pet ether) to give compound 12 as a white solid (26 mg, 76%): [α]25D −5.1 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.42 (d, J = 2.8 Hz, 1H, H-4), 5.36 (m, 1H, HC), 5.20 (t, J = 9.2 Hz, 1H, H-3′), 5.10 (dd, J = 10.0, 3.4 Hz, 1H, H-3′′), 5.05–4.97 (m, 3H, H-2′′, H-4′ & H-4′′), 4.90 (d, J = 1.3 Hz, 1H, H-1′′), 4.77 (dd, J = 10.3, 3.2 Hz, 1H, H-3), 4.65 (d, J = 7.4 Hz, 1H, H-1′), 4.44 (d, J = 7.6 Hz, 1H, H-1), 4.38 (dd, J = 12.3, 3.8 Hz, 1H, H-6a), 4.18–4.05 (m, 3H, H-6a′, H-6b′ & H-6b), 3.98–3.93 (m, 2H, H-2), 3.87 (t, J = 6.8 Hz, 1H, H-5), 3.72–3.67 (m, 1H, H-5′) 3.62 (t, J = 9.4 Hz, 1H, H-2′) 3.53–3.45 (m, 1H, H-5′′), 2.35–2.29 (m, 2H), 2.11 (s, 3H, CH3), 2.07 (s, 6H, 2CH3), 2.03 (s, 9H, 3CH3), 2.02 (s, 3H, CH3), 1.98 (s, 3H, CH3), 1.97 (s, 3H, CH3), 1.89–1.82 (m, 3H), 1.68–1.42 (m, 8H), 1.39–1.31 (m, 9H), 1.11 (d, J = 6.0 Hz, 3H, CH3), 1.18–1.07 (m, 8H), 1.01 (s, 3H), 0.94–0.93 (m, 9H), 0.68 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 170.6, 170.5, 170.4, 170.3, 170.2, 170.1, 169.8, 169.7, 133.6, 117.3, 101.7, 98.4, 98.1, 76.6, 74.4, 73.2, 71.7, 69.3, 69.0, 68.9, 68.3, 68.2, 66.9, 66.2, 61.8, 61.7, 20.95, 20.90, 20.84, 20.83, 20.78, 20.74, 19.5, 18.8, 17.5, 12.0; HRMS calcd for C63H94O24 [M + Na]+ 1257.6027, found 1257.6035.
Cholesteryl α-L-rhamnopyranosyl-(1→2)-β-D-glucopyranosyl-(1→2)-β-D-galactopyranoside (1c)
NaOMe (40 mg) was added to a clear solution of 12 (35 mg) in CH2Cl2:MeOH (4 mL, 1:1) and the reaction mixture was kept for stirring at rt for 8 h. After complete consumption of starting material, reaction mixture was neutralized with amberlite (acidic resin) and filtered. The filtrate was concentrated and chromatographed on silica gel (15% MeOH in EtOAc) to give the desired product 1c as white solid (20 mg, 86%): [α]20D −17.2 (c 0.57, MeOH); 1H NMR (500 MHz, MeOD) δ 5.39 (s, 1H), 5.25 (s, 1H), 4.89 (1H under MeOD), 4.43 (d, J = 7.8 Hz, 1H), 4.17–4.09 (m, 1H), 4.06–3.57 (m, 11H), 3.54–3.36 (m, 4H), 3.28–3.22 (m, 2H), 2.48–2.46 (m, 1H), 2.29–2.27 (m, 1H), 2.08–2.01 (m, 1H), 2.01–1.90 (m, 4H), 1.89–1.45 (m, 8H), 1.49–1.24 (m, 13H), 1.21–1.10 (m, 7H), 1.09–1.0.99 (m, 5H), 0.96 (d, J = 6.0 Hz, 3H), 0.92–0.82 (m, 6H), 0.72 (s, 3H); 13C NMR (125 MHz, MeOD) δ 142.1, 122.9, 102.2, 102.1, 81.4, 79.45, 79.40, 77.9, 77.3, 76.7, 76.0, 74.2, 72.4, 72.3, 70.9, 69.8, 69.3, 63.4, 62.6, 61.7, 58.3, 57.7, 43.6, 41.3, 40.8, 40.2, 38.6, 38.0, 37.5, 37.2, 33.4, 33.2, 30.9, 30.87, 30.82, 30.62, 30.60, 29.4, 29.2, 25.4, 25.0, 23.8, 23.3, 23.0, 22.3; HRMS calcd for C45H76O15 [M + Na]+879.5076, found 879.5071.
Acknowledgements
This work was supported by the Department of Science and Technology (grant no. SR/S1/OC-40/2009) and Council of Scientific and Industrial Research (grant no. 01(2376)/10/EMR-II). ME thanks CSIR-New Delhi for a fellowship.Notes and references
- B. Yu, Y. Zhang and P. Tang, Eur. J. Org. Chem., 2007, 5145–5161 CrossRef CAS
.
-
(a) C. Abbet, M. Neuburger, T. Wagner, M. Quitschau, M. Hamburger and O. Potterat, Org. Lett., 2011, 13, 1354–1357 CrossRef CAS PubMed
- C.-A. Tai, S. S. Kulkarni and S.-C. Hung, J. Org. Chem., 2003, 68, 8719–8722 CrossRef CAS PubMed
- L. Jiang and T.-H. Chan, Tetrahedron Lett., 1998, 39, 355–358 CrossRef CAS
- S. S. Kulkarni, Y.-H. Liu and S.-C. Hung, J. Org. Chem., 2005, 70, 2808–2811 CrossRef CAS PubMed
- E. Dubois and J.-M. Beau, Carbohydr. Res., 1992, 228, 103–120 CrossRef CAS
- T. Oka, K. Fujiwara and A. Murai, Tetrahedron, 1996, 52, 12091–12110 CrossRef CAS
- G. Catelani, F. Colonna and A. Marra, Carbohydr. Res., 1988, 182, 297–300 CrossRef CAS
- P. Wang, H. Lee, M. Fukuda and P. H. Seeberger, Chem. Commun., 2007, 1963–1965 RSC
- V. Pedretti, A. Veyrières and P. Sinaÿ, Tetrahedron, 1990, 46, 77–88 CrossRef CAS
-
(a) J. M. Lassaletta, M. Meichle, S. Weiler and R. R. Schmidt, J. Carbohydr. Chem., 1996, 15, 241–254 CrossRef CAS
- E. M. Scanlan, M. M. Mackeen, M. R. Wormald and B. G. Davis, J. Am. Chem. Soc., 2010, 132, 7238–7239 CrossRef CAS PubMed
- Y. Du, M. Zhang and F. Kong, Org. Lett., 2000, 2, 3797–3800 CrossRef CAS PubMed
- M. R. Pratt and C. R. Bertozzi, Org. Lett., 2004, 6, 2345–2348 CrossRef CAS PubMed
- P. I. Kitov and D. R. Bundle, Org. Lett., 2001, 3, 2835–2838 CrossRef CAS PubMed
- J. Gao, X. Li, G. Gu, B. Sun, M. Cui, M. Ji and H.-X. Lou, Bioorg. Med. Chem. Lett., 2011, 21, 622–627 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for all new compounds, and copies of 1H, 13C and 2D NMR spectra. See DOI: 10.1039/c3ra47523a |
| This journal is © The Royal Society of Chemistry 2014 |