
Candida antarctica lipase B-catalyzed synthesis of polyesters: starting from ketones via a tandem BVO/ROP process†
Abstract
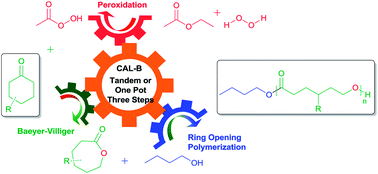
A novel enzymatic tandem procedure for the synthesis of substituted polyesters starting from ketones has been developed. Candida antarctica lipase B (CAL-B) was used as a catalyst for the whole procedure consisting of the Baeyer–Villiger oxidation (BVO) of substituted ketones and the subsequent ring-opening polymerization (ROP) of corresponding substituted lactones. The products of oxidation and polymerization were characterized by 1H-NMR, IR and GPC. The substituents of the ketones greatly influenced the molecular weights of the obtained polyesters, ranging from 1000 to 12 800.
 Please wait while we load your content...
Please wait while we load your content...

