 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Luminescence properties of europium titanate thin films grown by atomic layer deposition
Per-Anders
Hansen
*a,
Helmer
Fjellvåg
a,
Terje G.
Finstad
b and
Ola
Nilsen
a
aDepartment of Chemistry, Centre for Materials Science and Nanotechnology, University of Oslo, Sem Sælandsvei 26, 0371 Oslo, Norway. E-mail: p.a.hansen@kjemi.uio.no
bDepartment of Physics, Centre for Materials Science and Nanotechnology, University of Oslo, Sem Sælandsvei 24, 0371 Oslo, Norway
First published on 17th February 2014
Abstract
Luminescent europium titanium oxides (EuxTiyOz) have been deposited as thin films by atomic layer deposition (ALD) in the temperature range 225 to 375 °C with control of the stoichiometry from pure TiO2 to pure Eu2O3, and their luminescent properties have been characterized. The current work demonstrates the feasibility of tailoring the luminescent properties of these films by varying ALD deposition parameters. The highest luminescence efficiency was observed for the amorphous phase while it decreases steadily with increasing annealing temperature up to 700 °C due to the formation of crystalline Eu2Ti2O7. The limiting factor for luminescence at low Eu contents is the low concentration of emitting Eu3+ atoms, while the luminescence at high Eu contents is limited by a low UV absorption due to the low Ti4+ content in these samples. The precursors used for the Ti–O deposition were TiCl4 and H2O, while Eu(thd)3 (thd = 2,2,6,6-tetramethyl-3,5-heptanedione) and ozone were used for the deposition of Eu–O. The films have been characterized by spectroscopic ellipsometry, photoluminescence, X-ray diffraction, X-ray fluorescence and atomic force microscopy. The depositions gave smooth (RMS roughness < 0.4 nm) and amorphous films for 30% and more Eu–O cycles (9.5 cationic% Eu), while 10 and 20% gave rough and partly crystalline films. The refractive index, extinction coefficient and luminescence efficiency are shown to vary continuously with Eu content for 30% Eu–O cycles or more, while the shape of the luminescence spectrum is independent of Eu content in the same range. For the 10 and 20% Eu–O depositions, we observed a gradual change in the luminescence spectra, coinciding with an increase of the proportion of anatase phase with decreasing Eu content.
Introduction
Luminescent materials find many applications, including phosphors for lighting and TV screens, lasers, markers for security tags and in biology, optical amplifiers and for energy applications like solar cells. Silicon based solar cells1,2 as well as thin film silicon cells3 have been shown to benefit from thin film phosphor materials. Models show that in particular CdTe based solar cells would benefit from such films.4 For luminescent materials, the crystal structure, texture, purity, defects, distribution of atoms and in particular the site symmetry of the luminescent center affects the luminescence efficiency of the material. In general, large single crystals are preferable to polycrystalline samples in order to reduce the surface defects and scattering, while thin film processes is helpful to tailor the texture and distribution of dopants.Atomic layer deposition offers unique possibilities in designing new materials and thin films. The low temperature layer-by-layer growth allows nanostructuring and next-neighbor distribution of atoms not easily achievable by other methods. The low deposition temperatures often results in growth of amorphous materials. This is fortunate as it enables investigation of the correlations between the luminescence efficiency and the crystallinity, or lack thereof. Europium titanate was chosen as a model material in this work due to its luminescent properties and because it could be obtained as thin coatings by combining the known deposition processes of Eu2O3 and TiO2 using the Eu(thd)3/O3 (ref. 5 and 6) and TiCl4–H2O7 precursor pairs.
Europium titanate has previously been synthesized by several techniques and giving rise to several types of microstructures. The synthesis routes includes hydrothermal,8–10 sol–gel,11–20 high temperature firing,21 flame pyrolysis22 and electrospinning.23,24 The different types of microstructures includes hollow spheres,8 thin films,12,16,17,19,20 porous films,13,18 nanoparticles,9,15,22,25 nanofibers,23,24 nanotubes10 and powders.11,21 Europium titanium oxides represents a diverse and fairly well known system which make them suitable for investigating the effects that layered ALD-type growth has on their luminescent properties.
The current work focuses on the application of ALD for deposition of luminescent materials. First, we explore the luminescence properties of thin films of europium titanium oxides as a function of their europium content, ranging from pure TiO2 to pure Eu2O3. Secondly, we describe how annealing changes the crystallinity of as-deposited films and thereby the luminescence of the samples. Lastly, we investigate the correlation between the deposition temperature and the composition, crystallinity and luminescence properties of the films.
Experimental
The thin films were deposited in a F-120 Sat-type ALD-reactor (ASM Microchemistry Ltd) using a combination of the processes for deposition of Eu2O3,5 and TiO2.7 Eu2O3 was deposited using Eu(thd)3 [europium tris(2,2,6,6-tetramethyl-3,5-heptanedionate), Strem > 99.9%] and ozone, and TiO2 was deposited using TiCl4 (Aldrich > 99.9%) and water. The ozone was produced from 99.8% O2 (AGA) in a BMT 803N Ozone Generator. Nitrogen was used as carrier and purge gas, and was separated from air in a nitrogen generator (Schmidlin UHPN3001 N2 purifier, >99.999% N2 + Ar purity). The sublimation temperature used for Eu(thd)3 was 145 °C, and the pulse durations for the deposition of Eu2O3 layers were 1.5/1.5/4/1.5 s for Eu(thd)3/purge/O3/purge. For TiO2, pulse durations were 0.8/1/1/1 s for TiCl4/purge/H2O/purge. These pulse and purge times have through prior experience proven to be sufficient for proper surface saturation and purging for the reactor used, in agreement with prior work.5 The substrates used for all depositions were cut from polished Si(100) wafers into pieces of 3 × 3 cm2. One of these substrates was used per deposition. In addition, 2 strips of 0.5 × 4 cm2 were cut from the same type of wafer and positioned perpendicular to the gas flow and about 8 cm apart by the inlet and the exhaust of the deposition chamber to monitor the gradients of the growth. The native oxide on the silicon was not removed. All substrates were cleaned in ethanol and dried using pressurized air. The substrates were further pre-cleaned by an in situ 30 minute pulsing sequence of ozone and nitrogen at the deposition temperature to remove any organic residues prior to the deposition. To aid comparison, the total film thicknesses of the samples were kept at ca. 80 nm, based on the growth rates deduced for Eu2O3, TiO2, and samples with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Eu:Ti pulsing ratio. The total number of cation cycles used per deposition is summarized in Table 1. For brevity, a sample consisting of 20% Eu2O3 and 80% TiO2 cycles will be called 20Eu. All films were deposited at 300 °C, while films of 50Eu were additionally deposited at 225 and 375 °C, keeping the number of cycles the same.
:1 Eu:Ti pulsing ratio. The total number of cation cycles used per deposition is summarized in Table 1. For brevity, a sample consisting of 20% Eu2O3 and 80% TiO2 cycles will be called 20Eu. All films were deposited at 300 °C, while films of 50Eu were additionally deposited at 225 and 375 °C, keeping the number of cycles the same.
| N | 0 | 10 | 20 | 30 | 40 | 50 | 60 | 70 | 80 | 90 | 100 |
| # | 1000 | 1080 | 1095 | 1520 | 1740 | 1930 | 2075 | 2180 | 2245 | 2300 | 2383 |
The crystallinity and phase content of the deposited films were evaluated by means of a Siemens D5000 X-ray diffractometer using CuKα1 (nm) radiation from a Ge(111) monochromator. The chemical stoichiometry was measured by X-ray fluorescence on a Phillips PW2400 Spectrometer and interpreted with the Uniquant analysis software. Annealing was conducted in a rapid thermal processing oven (RTP) in vacuum for 10 minutes at a given temperature using a heating rate of 25 °C s−1. Film thickness and refractive index were determined with a spectroscopic ellipsometer (J. A. Woollam VASE) in the 250–1000 nm range. The Tauc–Lorentz formalism26,27 was used to model and parameterize the ellipsometry experimental data. These thicknesses were used to determine the growth rate per cation cycle (GPC). Surface morphology was characterized with a Park XE70 atomic force microscope (AFM) in tapping mode. Photoluminescence in the films were excited by a 325 nm CdHe laser and detected by an OceanOptics USB4000 photospectrometer in the 350–1000 nm range. Diffuse reflectance measurements were done with a Shimadzu UV-3600 photospectrometer with an integrating sphere.
Results
Fig. 1 shows the growth rates of the deposited films for samples with different pulsed stoichiometries. The variation in growth rate show local minima at 30Eu and 90Eu and a local maximum at 50Eu. The films were rather uniform with negligible gradients, except for 10Eu and 20Eu. Thus the measured growth rate for the inlet and exhaust monitor strips are shown for 10Eu and 20Eu, while the other runs showed no significant variations in the film thickness. It can be seen from Fig. 1 that the growth rate at the gas inlet side was substantially lower than that at the exhaust side for 10Eu and 20Eu. The three data points for 10Eu and 20Eu are from three separate depositions. The growth rate of pure TiO2 was 73.9 pm per cycle which is consistent with the values reported by Aarik et al.7 for ALD under similar conditions.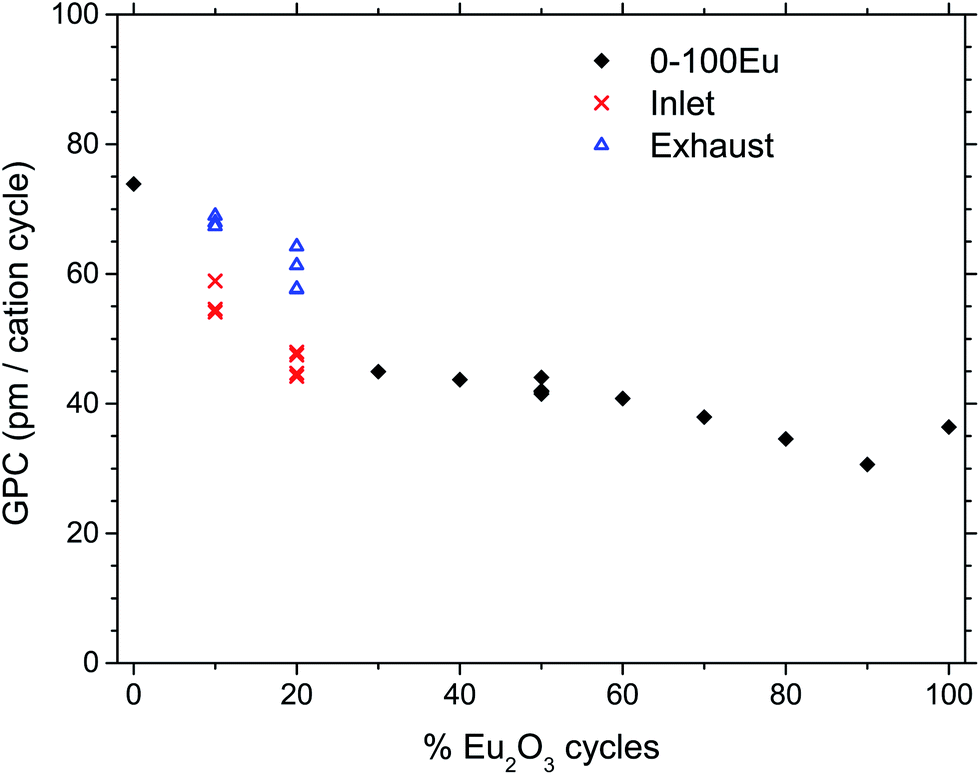 | ||
| Fig. 1 Growth rate per cation cycle (GPC) as a function of the percent of Eu2O3 cycles. Black diamonds indicate smooth depositions, while red and blue symbols indicate depositions with gradients, measured the inlet and exhaust of the reaction chamber, respectively. | ||
The dependency of the pulsing composition on the cationic composition of the deposited films was analyzed by X-ray fluorescence and showed a smooth variation as function of the percentage of Eu2O3 cycles, Fig. 2. The concentration of chlorine in the film was also measured to be mostly less that 0.5 at% except for 90Eu were the Cl concentration was nearly 2 at%.
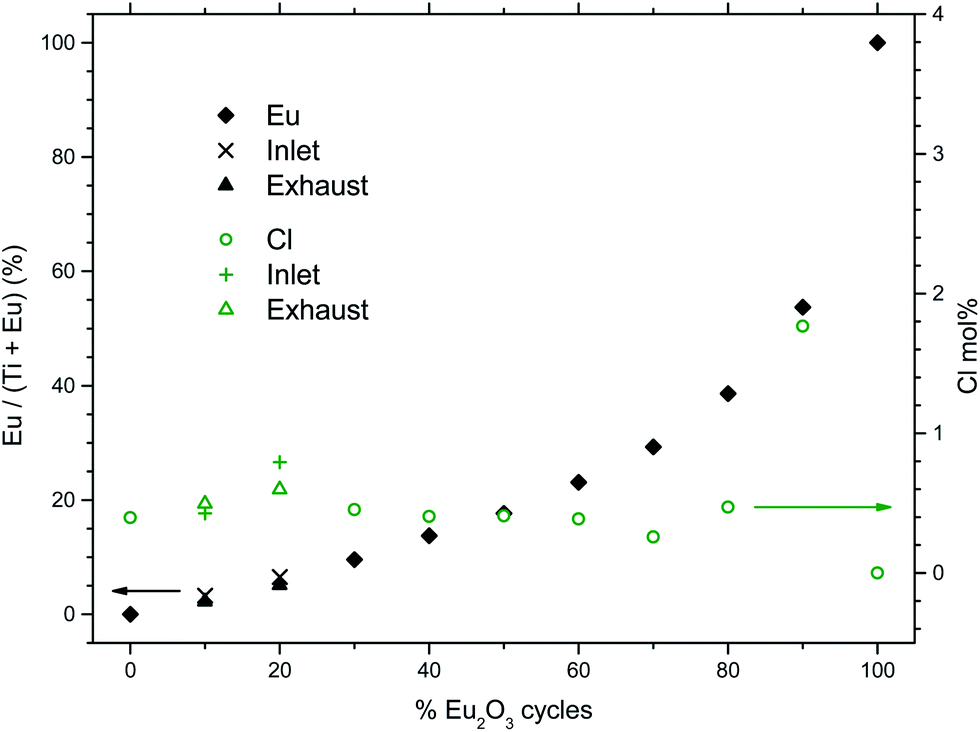 | ||
| Fig. 2 Cationic composition of the deposited films as function of % Eu2O3 cycles, as measured by XRF. The analyzed chlorine content is also shown (right scale). | ||
The crystallinity of the as-deposited films depended on the applied pulsing composition, as seen in Fig. 3. The X-ray diffractogram for 100Eu shows a broad feature around 30° that may stem from monoclinic Eu2O3,28 similar to what we found in our previous work on Ln2O3 depositions.5 For the range 30Eu to 90Eu, the depositions resulted in amorphous films. The TiO2 depositions (0Eu) gave polycrystalline anatase. For the range 10Eu to 20Eu, the growth characteristics depended on the position in the reaction chamber. The samples were consistently thinner at the gas inlet side than at the exhaust side. From the XRD data it is seen that the exhaust side contains more anatase than the inlet side for both 10Eu and 20Eu depositions. The expanded views of the (011) anatase reflection show that the peak is shifted from 25.4° for pure anatase to 25.3° for the Eu doped samples.
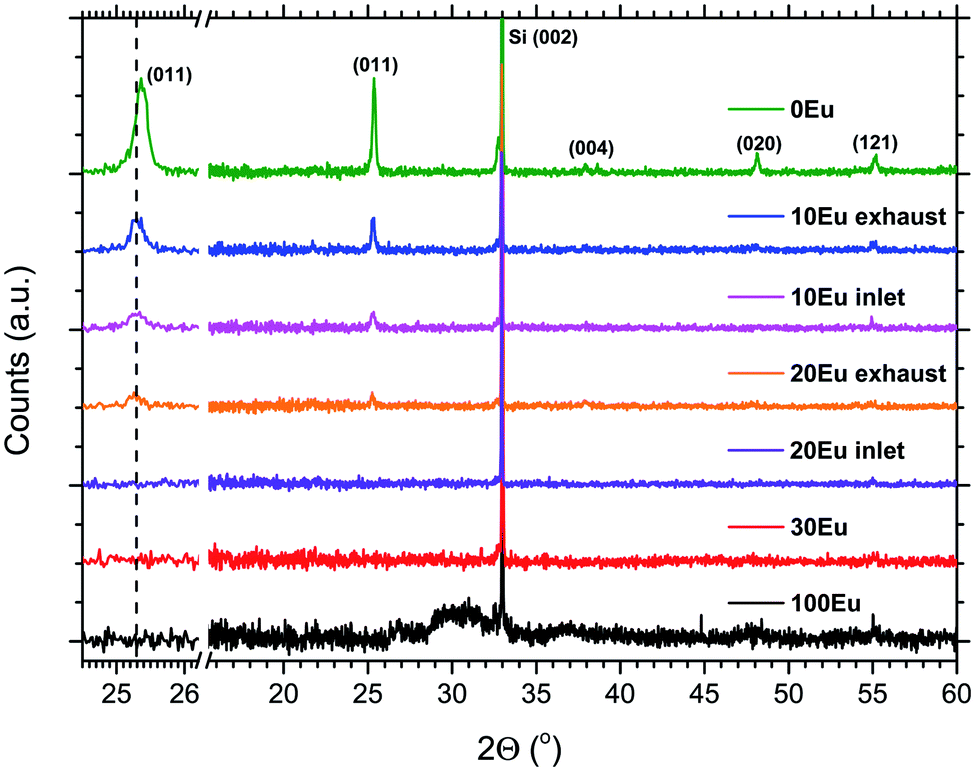 | ||
| Fig. 3 X-ray diffractograms for 0, 10, 20, 30 and 100% Eu2O3 cycles. The observed reflections are from the anatase polymorph of TiO2, while the sharp reflection at 33° is from the Si (001) substrate. The left side show an expanded view of the (011) anatase reflection. | ||
AFM images of the 10 and 20Eu samples are shown in Fig. 4. At the exhaust side, both samples consist of a dense layer of roughly 100 nm size crystallites. At the gas inlet, the crystallites are also ca. 100 nm in size, but the crystallite density is notably lower, in particular for the 20Eu sample where the crystallites are separated by smooth regions. The RMS roughness for the 3 × 3 μm2 area is 4.3 nm for 0Eu (not shown), 11 and 10 nm for 10Eu inlet and exhaust respectively, and 3.2 and 6.8 nm for 20Eu at the inlet and exhaust respectively. The smooth region between the crystallites has a RMS roughness of 0.1–0.3 nm, which also applies for the 30–90Eu samples. The 100Eu sample has a RMS roughness of 0.6 nm.
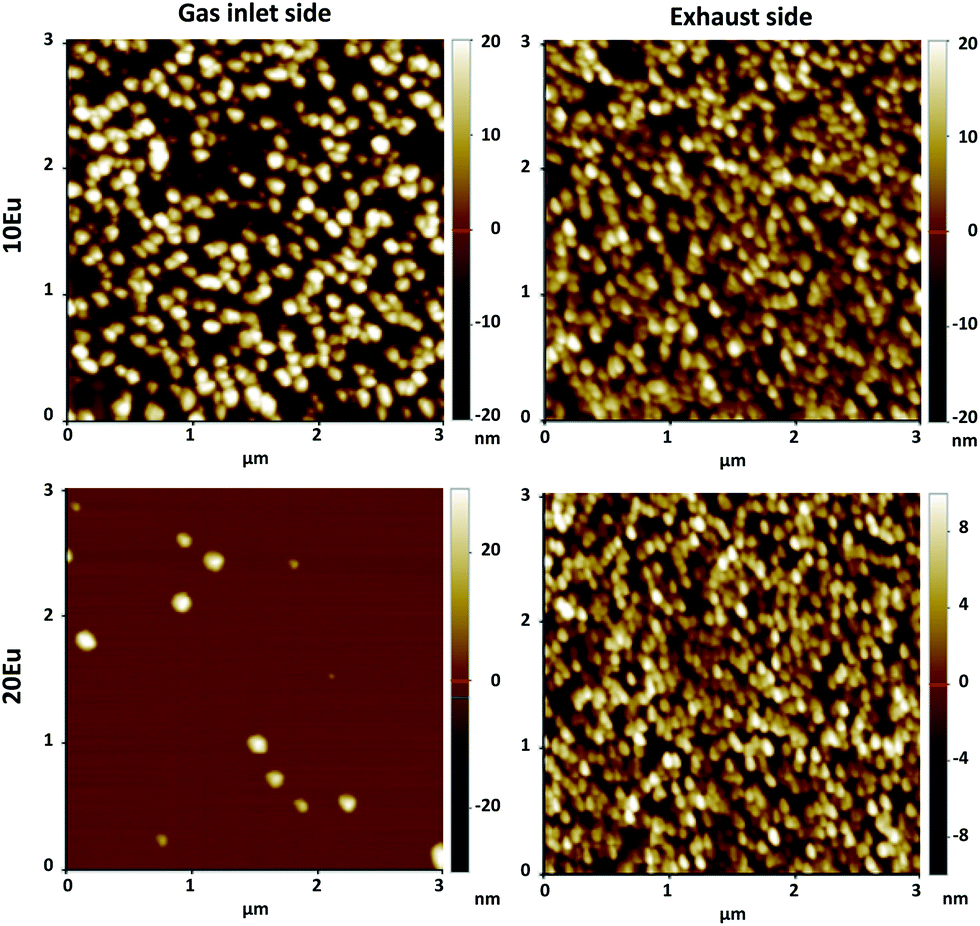 | ||
| Fig. 4 AFM pictures of 10Eu and 20Eu films at the inlet and exhaust side. Note the differences in the z scale. | ||
The PL spectra of the 10 to 90Eu depositions are presented in Fig. 5. The spectra are normalized to the most intense peak and the 0Eu spectrum is in addition multiplied by 0.5 for graphical reasons. The spectrum consists of a number of distinct lines. For Eu3+ they range from 580 to 710 nm, and are associated with the transitions from the excited state 5D0 to 7FJ (J = 0, 1, 2, 3 and 4) levels as indicated in the figure. The shape of the PL spectra is similar for the 30Eu to 90Eu depositions, in addition to the gas inlet side of the 20Eu depositions. There is a gradual change in the shape of the spectra going from the exhaust side of 20Eu to 10Eu inlet to 10Eu exhaust. This is particularly visible for the hypersensitive 5D0 → 7F2 transition.
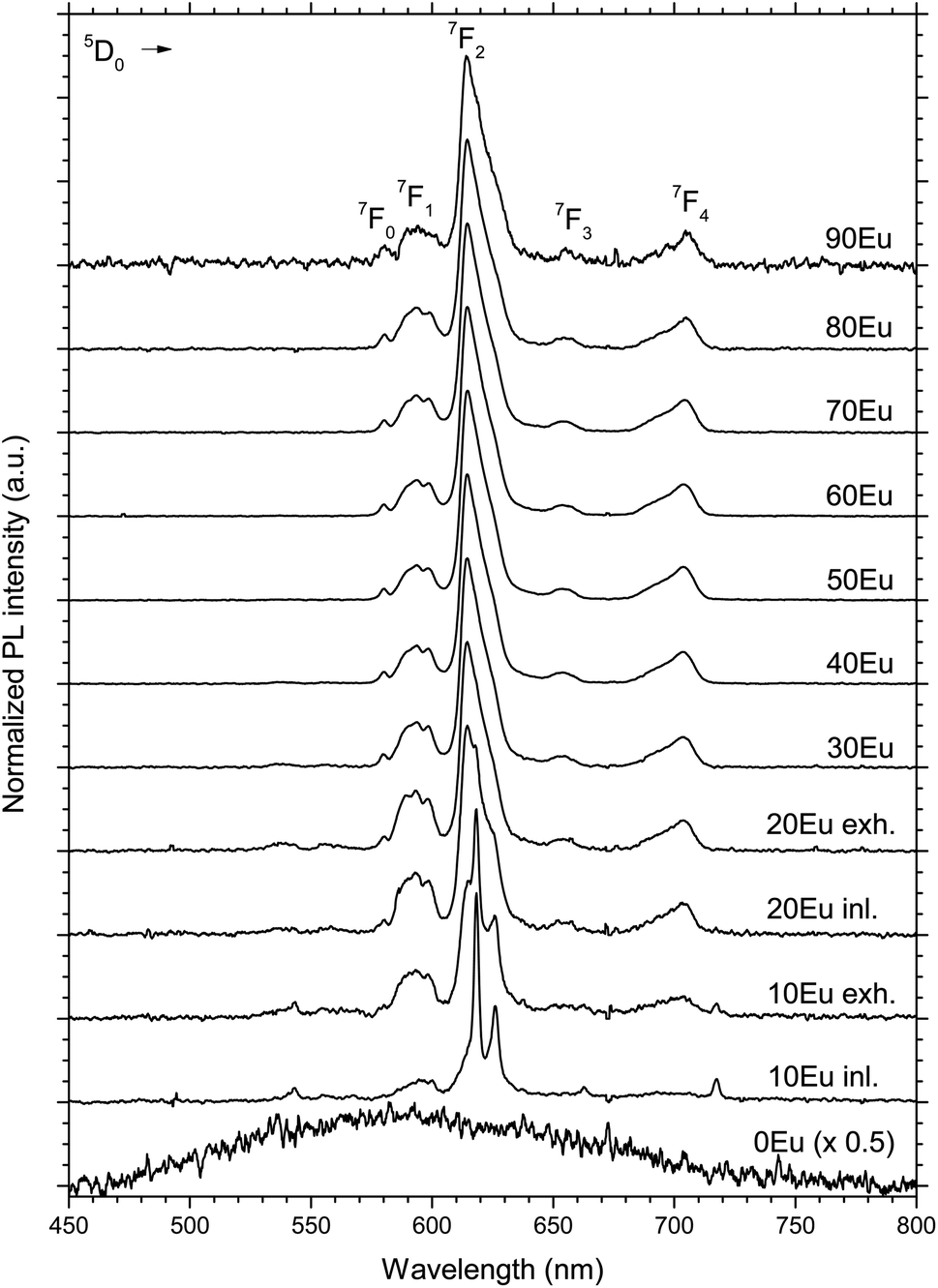 | ||
| Fig. 5 Normalized emission PL spectra of 0 to 90Eu samples, excited by a 325 nm HeCd laser. For the 10 and 20Eu samples, the inlet and exhaust sides are named “inl.” and “exh.” respectively. Some detector artifacts have been removed from the data and the 0Eu data have been multiplied by 0.5. | ||
The total PL intensity in the 550–750 nm range (400–900 nm used for 0Eu) varies with the europium content as shown in Fig. 6.
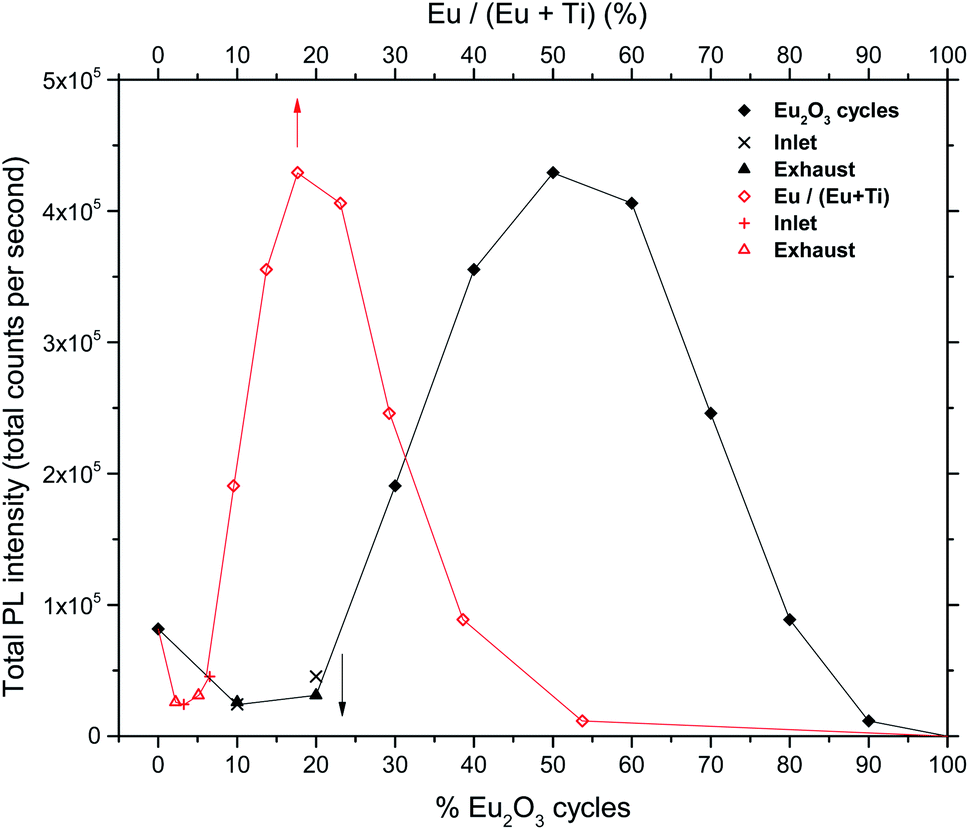 | ||
| Fig. 6 Total PL intensities of europium titanate films vs. cycle% Eu2O3 (black curve) and cation% Eu (red curve). | ||
In order to shed light on the luminescent properties of the samples, the extinction coefficient and refractive index was modeled from ellipsometry data, Fig. 7. The stapled line in Fig. 7 show the wavelength of the CdHe laser used for PL excitation. The refractive index (n) and extinction coefficient (k) at 325 nm as a function of Eu2O3 cycle ratio is given in Fig. 8. The extinction coefficient at the laser wavelength decreases with increasing Eu concentration down to zero for pure Eu2O3, while n decreases from 3.5 for TiO2 to 2.1 for Eu2O3. Variations in optical scattering between the samples can also cause differences in the measured emission intensities. Diffuse reflectance measurements at zero degree incidence showed that the scattering at both the excitation and emission wavelengths were similar for all the samples and below 1%, with the exception of the 10Eu and 20Eu samples which showed a diffuse reflectance of 2 and 1.2% respectively at 325 nm.
 | ||
| Fig. 7 Extinction coefficient from modeling VASE data, showing that k increases with decreasing Eu concentration. The 10Eu inlet sample was difficult to model in the UV range, probably due to its large surface roughness compared to the other samples. | ||
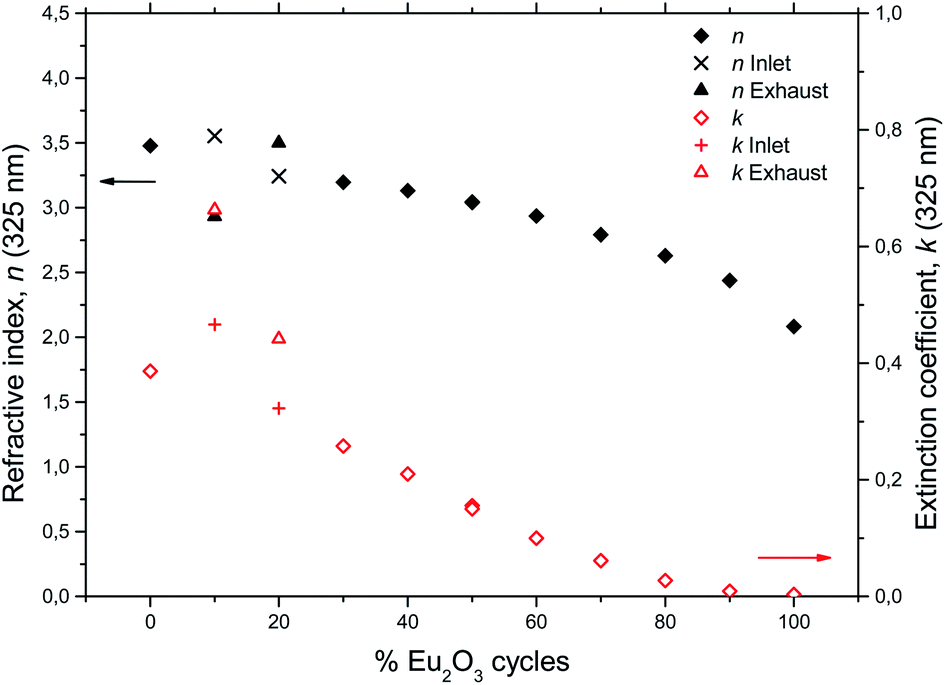 | ||
| Fig. 8 Refractive index and extinction coefficient at 325 nm for the europium titanium oxide depositions. | ||
The 50Eu depositions were in addition carried out at 225 °C and 375 °C, keeping a constant number of 965 supercycles (1 Eu2O3 + 1 TiO2 cycle), consistent with Table 1. These depositions also resulted in amorphous films similar to the depositions at 300 °C, as shown in Table 2. However, there are notable differences. The growth rate first increases and reaches a plateau at 300 °C, while the Eu concentration and total PL intensity increases and k decreases for increasing deposition temperatures.
| 225 °C | 300 °C | 375 °C | |
|---|---|---|---|
| Film thickness (nm) | 64.0 | 85.1 | 80.3 |
| GPC (pm per cation cycle) | 33.1 | 44.1 | 41.6 |
| Eu cat.% | 16.6% | 17.7% | 20.9% |
| Cl mol% | 1.3% | 0.4% | 0.3% |
| Total PL intensity (Counts) | 1.5 × 105 | 4.6 × 105 | 5.1 × 105 |
| n (325 nm) | 3.00 | 3.04 | 2.98 |
| k (325 nm) | 0.16 | 0.15 | 0.11 |
The 50Eu samples obtained at the three deposition temperatures were annealed in order to investigate the luminescence properties as function of film crystallinity. The films remain amorphous up to an annealing temperature of 800 °C where rutile starts to form, as can be seen in Fig. 9. After 900 °C, the (222)-reflection of Eu2Ti2O7 appeared, while after 1000 °C the sample is best described as a polycrystalline mixture of Eu2Ti2O7 and rutile.
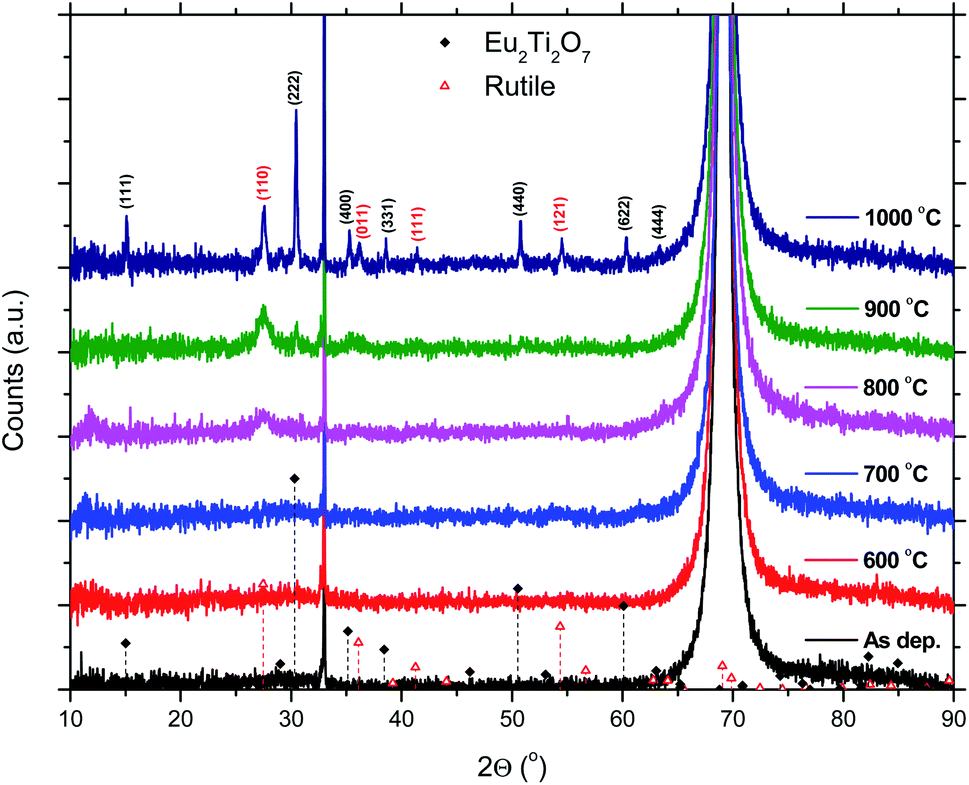 | ||
| Fig. 9 XRD data for a 50Eu films after annealing. The sharp reflection at 33° and the large reflection at 69° are from the Si(001) substrate. | ||
The total PL intensity decreased when 50Eu samples were annealed, Fig. 10. For annealing temperatures of 700 °C and above, the samples show very low luminescence. The exception is the sample deposited at 375 °C which show a higher luminescence when annealed at 800 °C than at 700 °C. The PL spectra for the samples deposited at 375 °C are shown in Fig. 11. The spectra are normalized to the highest peak. It is further seen from the figure that although the PL intensity decreases with annealing, the shape of the spectra remain similar for annealing temperatures up to 700 °C, whereafter it changed in shape.
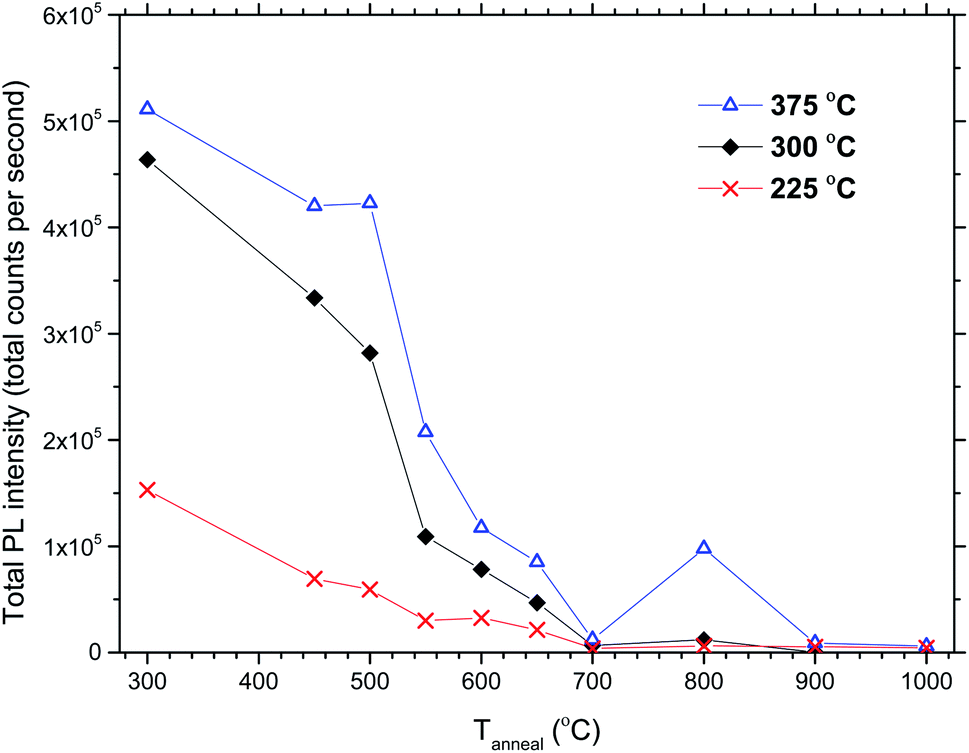 | ||
| Fig. 10 Total PL intensities vs. annealing temperature for 50Eu films deposited at 225, 300 and 375 °C. | ||
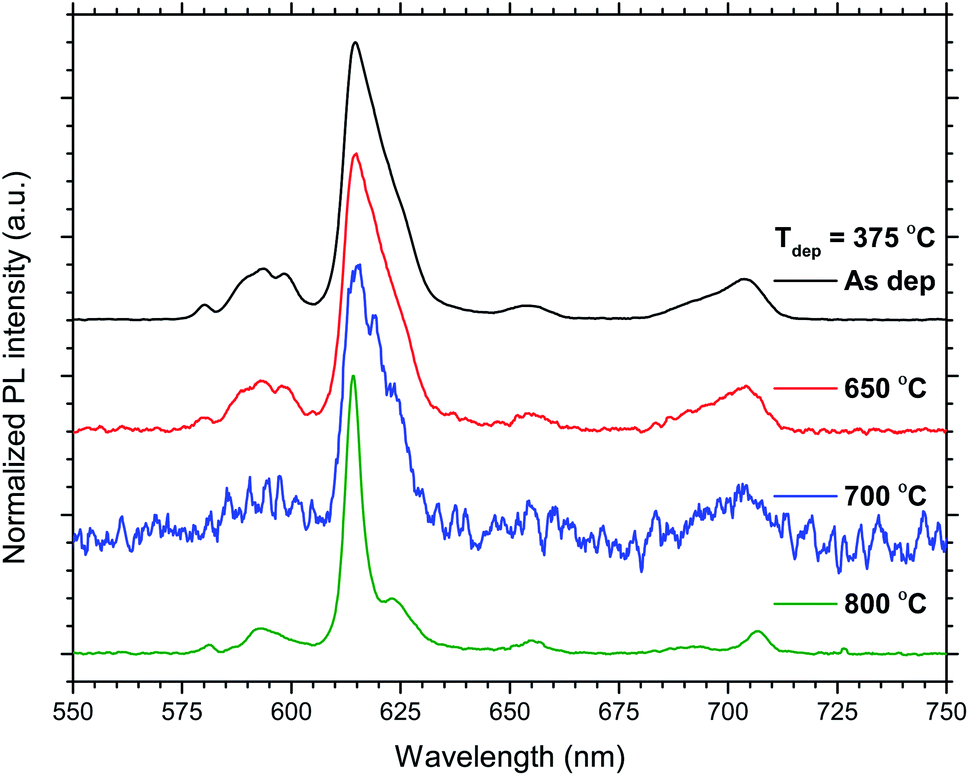 | ||
| Fig. 11 Normalized emission PL spectra of the 50Eu samples deposited at Tdep = 375 °C and the indicated temperatures. | ||
Discussion
By varying the overall composition of the deposited films between TiO2 and Eu2O3, several different phases were formed. From low to high Eu content, these phases were: anatase, europium doped anatase, amorphous EuxTiyOz, and poorly crystalline Eu2O3. The results further show that upon annealing the 50Eu samples at 800 °C and higher, rutile and crystalline Eu2Ti2O7 is formed from the amorphous oxide. In this work, XRD and PL have been used to distinguish these phases.The total emission intensity of our samples change with different Eu3+ concentration, deposition and annealing temperature. We have tried to design the samples to be optically similar to keep the main differences within k(λ), Eu3+ concentration, and their local symmetry. All samples were approximately of the same thickness and with a surface which is smooth and similar, as measured by AFM. In addition, diffuse reflectance measurements show that the scattering in the samples is rather low.
The Eu3+ PL is sensitive to the coordination and local environment around the cation site based on the fact that the splittings of the 2S+1LJ multiplets and the transition probabilities between them are symmetry dependent. Usually the transition 5D0 → 7F2 is the strongest. It is hypersensitive to site symmetry and of electric-dipole nature. For comparison, the 5D0 → 7F1 transition is mostly of magnetic-dipole nature and insensitive to site symmetry.29 The current discussion is divided into three parts: anatase containing 0Eu to 20Eu samples, amorphous 30Eu to 100Eu samples, and annealed 50Eu samples.
0Eu to 20Eu: anatase and EuxTiyOz
The 10 and 20Eu samples showed a notable difference in growth positioned at the inlet and exhaust side of the reaction chamber (Fig. 1). The films grown on the monitor strips at the inlet were thinner than those at the exhaust. The entire substrates appeared either thin or thick rather than exhibiting a smooth gradient through the chamber. This growth pattern was reproducible. The thinner films have a lower Ti content (Fig. 2). The AFM images (Fig. 4) show that the size of the crystallites is similar for all the samples. However, there are differences with respect to the density of crystallites, indicating that the nucleation of crystallites in the amorphous matrix is a limiting process.The PL spectra in Fig. 5 suggests that these films consist of two distinct phases as they can be regarded as the superposition of two characteristic PL spectra; the spectra of 80Eu having broad peaks from the 7FJ manifolds, and the sharp lines of the inlet strip of 10Eu. The superposition and gradual growth of one and decay of the other phase can be observed most clearly for the 7F2 peak. The PL from the inlet strip of 10Eu also has a shape that is consistent with Eu3+ doped anatase.13,18 The PL from 80Eu has smeared out peaks which one can imagine arise from the continuous slight variation in the coordination environment for a cation in an amorphous oxide where the bond angles are varying. Very similarly shaped PL spectra have also been observed before and been described as typical for a Eu3+ doped disordered system.10 Leroy et al.13 observed very similar spectra for PL from Eu3+ doped mesoporous titania with the same gradual spectrum change accompanying the change from anatase to amorphous phase.
Comparing the AFM images, XRD data, and PL spectra, there is a clear correlation between the amount of crystallites, the XRD peak intensities, and ratio between the two overlapping PL spectra. This correlation only applies for samples with less than 30% Eu2O3 cycles. Any correlation between the extinction coefficient k and ratio between the anatase and amorphous phases is less clear. For example, at the PL excitation wavelength of 325 nm the extinction coefficient does not increase monotonically with decreasing %Eu below 30Eu. This lack of correlation for k can also have to do with difficulties of adopting a reliable optical model for the ellipsometry data used to arrive at k for this range of %Eu and wavelength. Since the size of the crystallites is close to the wavelength of the light in the UV part of the spectrum, a lot of scattering is expected for these samples. Such scattering is not trivial to model and could result in some uncertainty in k in the UV range of these samples. Scattering in the UV range does not affect the modeling of thicknesses as this is done by modeling the VIS and NIR range ellipsometry data.
By comparing the (011) reflection of pure and doped anatase in Fig. 3, the peak shift from 25.4 to 25.3° indicates a slight unit cell expansion on incorporation of the larger Eu3+ ions. The shift is identical for both 10Eu and 20Eu, indicating that the anatase solid solution phase is likely to be saturated with Eu3+ in all these samples. Thus, for low Eu contents, simultaneous growth of amorphous EuxTiyOz and Eu-saturated anatase may occur. This competition between the growths of the two phases may explain the difference in growth rate between the samples positioned at the inlet and exhaust side in the reaction chamber.
30Eu to 100Eu: EuxTiyOz
The depositions in the 30Eu to 100Eu range all resulted in amorphous and very flat films with a RMS roughness of approximately 0.3 nm, with the exception of pure Eu2O3 (100Eu) which show sign of poor crystallinity (Fig. 3) and a slightly higher roughness of 0.6 nm. The normalized PL spectra in Fig. 5 indicate that the coordination of the Eu3+ is similar for all of these samples. On the other hand, the emission intensity varies strongly with the Eu content as shown in Fig. 6. At low Eu content, the luminescence intensity increases with the Eu content. The limiting factor for the emission in this Eu range is likely the energy transfer from TiO2 to Eu3+. Increasing the Eu content increases the likelihood of an Eu3+ being available to accept the energy transfer thus increasing the emission intensity. The strongest emission is seen for the 50Eu sample which corresponds to 17.7 cat.% Eu. At higher Eu contents, the luminescence intensity decreases.Although it would be tempting to assume that the decrease in luminescence intensity for >50Eu samples is caused by concentration quenching, we do not believe this is the case. At least part of the reason for the decrease in emission intensity is that k (325 nm) decreases, as seen in Fig. 7. This leads to a reduction in the absorption of the 325 nm laser in the thin films. Ovenstone et al.14 found that for amorphous EuxTiyOz powders, the emission intensity increased continuously for x up to 0.5 (compared to our maximum at x = 0.177), which was the highest Eu content in their investigation. This supports our explanation about the lack of laser absorption being the limiting factor for our thin films with high Eu contents, as UV absorption will not be a limiting factor in bulk powders. If thicker films had been grown, the emission maximum would likely be at higher Eu contents as more of the laser would be absorbed. For pure Eu2O3, the emission was not detectable with our set-up and only a very weak red spot was visible by eye, as the absorption at 325 nm is close to zero.
Effects of the deposition temperature and annealing on EuxTiyOz
The effects resulting from different deposition temperatures are summarized in Table 2. The temperature dependence of the TiCl4–H2O and Eu(thd)3/O3 ALD growth processes may explain these results. It is known that the growth rate of TiO2 decreases7 while that of Eu2O3 increases5 with temperature. Thus, by keeping the cycle sequence the same, one would expect a relative increase in the Eu content with increasing deposition temperature, which is consistent with the XRF results. Also, the extinction coefficient decreases with the relative decrease in the Ti content at higher temperatures. Still, the change in emission intensity cannot be fully explained by the growth rates. As the number of cycles was constant for these samples, the lower growth rate at 225 °C resulted in a thinner film, but not sufficiently thin to explain why the emission intensity should be reduced to just 1/3 of that of the 300 °C deposited sample. Possibly, the higher Cl content in the sample grown at 225 °C could indicate that the surface reactions during the ALD growth is more incomplete at this temperature than at higher temperatures.Since the 50Eu samples showed the highest emission intensity, we chose to anneal these samples to investigate how changes in the amorphous nature would affect the luminescence properties of these films. Fig. 9 shows that the films remained amorphous for annealing temperatures up to 800 °C, whereafter rutile started to crystallize. The emission intensity decreases with increasing annealing temperature, as seen in Fig. 10, and remains low for all annealing temperatures above 700 °C. This is similar to what Ovenstone et al.11 found for amorphous europium titanate powders. However, their longer annealing times gave a lower quenching temperature. The only exception currently observed is the deposition at 375 °C, for which the emission intensity is higher after annealing at 800 °C annealing than at 700 °C. The origin of this is unclear. Whereas the XRD analysis could not reveal any crystalline phases for these samples, the PL spectra in Fig. 11 indicates that the coordination environment of the Eu3+ ions changes. However, at this point we do not have sufficient experimental data to indicate what kind of coordination or what kind of crystal phase this spectrum corresponds to. A change in PL spectrum after annealing at 800 °C was also observed for the depositions at 225 and 300 °C, but these samples did not experience an increase in emission intensity and the luminescence intensity was very low.
Conclusion
By using the precursor pairs TiCl4–H2O and Eu(thd)3/O3 in ALD growth processes it is possible to obtain controlled Ti–Eu oxide materials by varying the cycle ratio, pulse duration and annealing temperatures. The observed phases are anatase, europium doped anatase, amorphous EuxTiyOz, crystalline Eu2Ti2O7 and poorly crystalline Eu2O3. The highest luminescence emission intensity was found for amorphous samples grown with alternating Eu2O3 and TiO2 cycles. The limiting factor for luminescence at low Eu contents is the low concentration of emitting Eu3+ atoms, while the luminescence at high Eu contents is limited by a low UV absorption due to the low Ti4+ contents in these samples.From a general point of view it is interesting to note that large crystals are in fact not always preferable. We were indeed expecting better luminescence efficiency after annealing due to formation of larger crystals, but rather observed that the amorphous phase gave the highest luminescence efficiency. From a practical point of view, this can be rather advantageous. Large single crystalline films are usually only obtainable through epitaxial growth on single crystal substrates, while amorphous films can be achieved through ALD-growth on most types of substrates.
Acknowledgements
This work was performed within ”The Norwegian Research Centre for Solar Cell Technology” project number 193829, a Centre for Environment-friendly Energy Research co-sponsored by the Research Council of Norway and research and industry partners in Norway. The authors would also like to thank the Department of Geosciences, UiO, for use of the XRF equipment, and the Institute for Energy Technology for use of the VASE ellipsometer.References
- T. Jin, S. Inoue, K. i. Machida and G. y. Adachi, J. Electrochem. Soc., 1997, 144, 4054–4058 CrossRef CAS PubMed.
- V. Švrček, A. Slaoui and J. C. Muller, Thin Solid Films, 2004, 451–452, 384–388 CrossRef PubMed.
- B.-C. Hong and K. Kawano, Sol. Energy Mater. Sol. Cells, 2003, 80, 417–432 CrossRef CAS PubMed.
- E. Klampaftis, D. Ross, K. R. McIntosh and B. S. Richards, Sol. Energy Mater. Sol. Cells, 2009, 93, 1182–1194 CrossRef CAS PubMed.
- P.-A. Hansen, H. Fjellvag, T. Finstad and O. Nilsen, Dalton Trans., 2013, 42(30), 10778–10785 RSC.
- J. Päiväsaari, M. Putkonen and L. Niinistö, Thin Solid Films, 2005, 472, 275–281 CrossRef PubMed.
- J. Aarik, A. Aidla, T. Uustare and V. Sammelselg, J. Cryst. Growth, 1995, 148, 268–275 CrossRef CAS.
- X. Feng, L. Yang, N. Zhang and Y. Liu, J. Alloys Compd., 2010, 506, 728–733 CrossRef CAS PubMed.
- H. Li, K. Zheng, X. Xu, H. Zhao, Y. Song, Y. Sheng, Q. Huo and H. Zou, Powder Technol., 2012, 228, 277–283 CrossRef CAS PubMed.
- P. Haro-González, M. Pedroni, F. Piccinelli, L. L. Martín, S. Polizzi, M. Giarola, G. Mariotto, A. Speghini, M. Bettinelli and I. R. Martín, J. Lumin., 2011, 131, 2473–2477 CrossRef PubMed.
- J. Ovenstone, P. J. Titler, R. Withnall and J. Silver, J. Phys. Chem. B, 2001, 105, 7170–7177 CrossRef CAS.
- L. A. Rocha, E. F. Molina, K. J. Ciuffi, P. S. Calefi and E. J. Nassar, Mater. Chem. Phys., 2007, 101, 238–241 CrossRef CAS PubMed.
- C. M. Leroy, T. Cardinal, V. Jubera, M. Treguer-Delapierre, J. Majimel, J. P. Manaud, R. Backov, C. Boissière, D. Grosso, C. Sanchez, B. Viana and F. Pellé, ChemPhysChem, 2008, 9, 2077–2084 CrossRef CAS PubMed.
- J. Ovenstone, P. J. Titler, R. Withnall and J. Silver, J. Mater. Res., 2002, 17, 2524–2531 CrossRef CAS.
- J. Beatriz, C. Rosa, C. Eloisa, E. Purificación, V. Bruno and S. Clément, Nanotechnology, 2005, 16, 2707 CrossRef.
- N. V. Gaponenko, I. S. Molchan, G. E. Thompson, P. Skeldon, A. Pakes, R. Kudrawiec, L. Bryja and J. Misiewicz, Sens. Actuators, A, 2002, 99, 71–73 CrossRef CAS.
- R. Palomino-Merino, A. Conde-Gallardo, M. Garcia-Rocha, I. Hernández-Calderón, V. Castaño and R. Rodriguez, Thin Solid Films, 2001, 401, 118–123 CrossRef CAS.
- C. Leroy, T. Cardinal, V. Jubera, M. Treguer-Delapierre, R. Backov, C. Boissière, D. Grosso, C. Sanchez, B. Viana and F. Pellé, J. Lumin., 2009, 129, 1641–1645 CrossRef CAS PubMed.
- A. Nebatti, C. Pflitsch, C. Eckert and B. Atakan, Prog. Org. Coat., 2010, 67, 356–360 CrossRef CAS PubMed.
- C. W. Jia, E. Q. Xie, J. G. Zhao, Z. W. Sun and A. H. Peng, J. Appl. Phys., 2006, 100, 023529 CrossRef PubMed.
- P. A. M. Berdowski and G. Blasse, J. Solid State Chem., 1986, 62, 317–327 CrossRef CAS.
- J.-G. Li, X. Wang, K. Watanabe and T. Ishigaki, J. Phys. Chem. B, 2005, 110, 1121–1127 CrossRef PubMed.
- J. Zhao, C. Jia, H. Duan, Z. Sun, X. Wang and E. Xie, J. Alloys Compd., 2008, 455, 497–500 CrossRef CAS PubMed.
- I. Cacciotti, A. Bianco, G. Pezzotti and G. Gusmano, Chem. Eng. J., 2011, 166, 751–764 CrossRef CAS PubMed.
- R. S. Ningthoujam, V. Sudarsan, R. K. Vatsa, R. M. Kadam, Jagannath and A. Gupta, J. Alloys Compd., 2009, 486, 864–870 CrossRef CAS PubMed.
- J. G. E. Jellison and F. A. Modine, Appl. Phys. Lett., 1996, 69, 371–373 CrossRef PubMed.
- J. G. E. Jellison and F. A. Modine, Appl. Phys. Lett., 1996, 69, 2137 CrossRef PubMed.
- G. Chen, J. R. Peterson and K. E. Brister, J. Solid State Chem., 1994, 111, 437–439 CrossRef CAS.
- J.-C. Bünzli and S. Eliseeva, in Lanthanide Luminescence, ed. P. Hänninen and H. Härmä, Springer, Berlin, Heidelberg, 2011, vol. 7, pp. 1–45 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |