
Total synthesis of phenanthroindolizidine alkaloids via asymmetric deprotonation of N-Boc-pyrrolidine†
Abstract
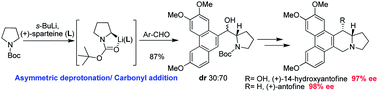
A concise and efficient enantioselective strategy to synthesize two typical phenanthroindolizidine alkaloids, 14-hydroxyantofine and antofine, was developed, featuring an asymmetric deprotonation/diastereoselective carbonyl addition sequence during which the formation of a chiral C-13a center and connection of pyrrolidine and phenanthrene moieties were achieved efficiently in one step. The absolute configuration of the C-13a stereocenter can be delicately controlled by using different enantiomers of sparteine, both of which are commercially available.
 Please wait while we load your content...
Please wait while we load your content...

