Oxazoline derivatives tagged with tosylated amino acids as recyclable organocatalysts for enantioselective allylation of aldehydes†
Debashis Ghoshab,
Arghya Sadhukhana,
Nabin Ch. Maitya,
Sayed H. R. Abdi*ab,
Noor-ul H. Khanab,
Rukhsana I. Kureshyab and
Hari C. Bajajab
aDiscipline of Inorganic Materials and Catalysis, CSIR-Central Salt and Marine Chemicals Research Institute (CSIR-CSMCRI), G. B. Marg, Bhavnagar-364 021, Gujarat, India. E-mail: shrabdi@csmcri.org; Fax: +91-0278-2566970
bAcademy of Scientific and Innovative Research, CSIR-Central Salt and Marine Chemicals Research Institute (CSIR-CSMCRI), Council of Scientific & Industrial Research (CSIR), G. B. Marg, Bhavnagar-364 021, Gujarat, India
First published on 17th January 2014
Abstract
A series of amino acid-based oxazoline compounds have been prepared and successfully applied to the enantioselective allylation reaction of aldehydes. The fine-tuning of the structure of the oxazolines led to (S,S)-4 as an efficient organocatalyst which gave homoallyl alcohols in good yield (up to 90%) and excellent ee (up to 99%) for a wide range of substrates including aromatic, hetero-aromatic and α,β-unsaturated aldehydes. The chiral organocatalyst was synthesized in three easy steps with an overall 88% yield and successfully recycled for up to three cycles. On the basis of the experimental observations and NMR studies, a probable mechanism was proposed for this reaction.
Introduction
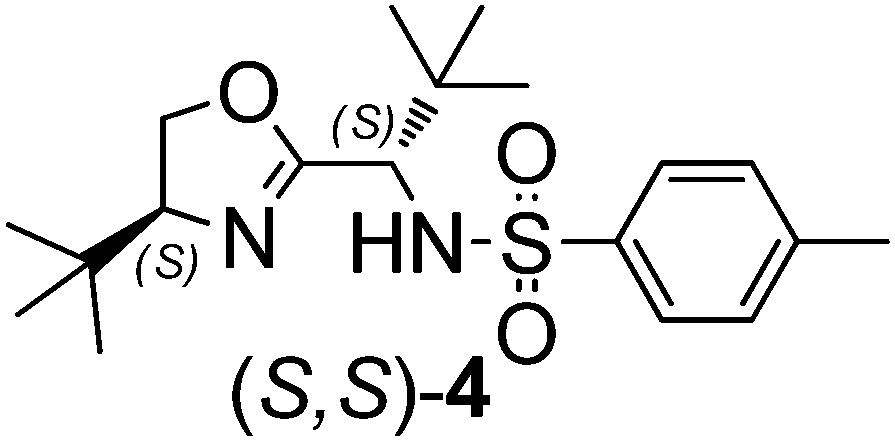
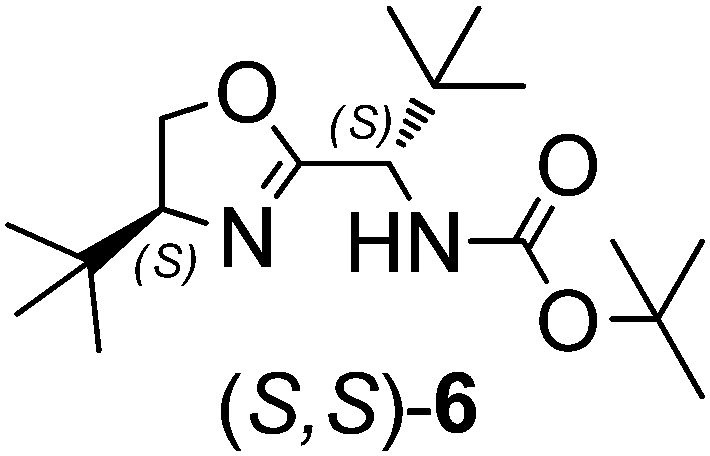
The application of organocatalysts in asymmetric C–C bond formation reactions, especially in asymmetric allylation reactions, is often preferred over metal-based catalysts, particularly in the pharmaceutical sector due to health, environmental, cost and efficiency concerns. For the development of new organocatalysts in asymmetric allylation reactions, various natural and synthetic amino acids as chiral building blocks seem to be most suitable due to their cost-effectiveness and easy availability. Chiral allylation products, i.e. homoallyl alcohols and homoallyl amines, are important building blocks for the construction of many biologically active compounds.1,2 For the production of homoallyl alcohol, allyltrialkylsilanes2a,c and stannanes3 react readily with aldehydes upon the activation of the carbonyl group by a Lewis acid.2,3 By contrast, allyltrichlorosilane requires activation with a Lewis base that coordinates to the silicon atom. Lewis bases reported in this respect are N-oxides,4 formamides,5 bisformamides,6 phosphine oxide,7 phosphinamides,8 urea derivatives9 and catecholates.10 Oxazoline derivatives as efficient ligands in metal-catalyzed asymmetric allylation reactions has been well documented,2c,11,12 however, Barrett et al.13 (1997) for the first time demonstrated that bidentate pyridine-linked oxazoline compounds can directly be used as organocatalysts for the allylation of aldehydes. Subsequently, chiral oxazolines appended with chiral sulfoxide14 and N-oxides4k have also been registered for their potential as organocatalysts for the asymmetric allylation of aldehydes. However, there are nagging issues, such as the extremely low reaction temperatures, moderate yield and ee, and non-recyclability of these catalysts, which need to be addressed. Bearing in mind the value of oxazolines, overall stability of the catalyst and our previous experience with tosylated amino acids15 as organocatalysts in the enantioselective allylation reaction of aldehydes, herein we have synthesized a series of oxazoline-based organocatalysts,16,17 (S,S)-1 to (S,S,R)-5, featuring sulfonamide groups with varying steric features and two to three chiral centers. Chiral centers in organocatalysts featuring configurations with different permutations and combinations were prepared for their possible role in influencing the product enantioselectivity. In order to know the specific role of the sulfonamide moiety in the organocatalyst on the activity and enantioselectivity of the allylation reaction, we also synthesized a Boc-protected organocatalyst, (S,S)-6.18 Among these organocatalysts, (S,S)-4 (10 mol%) was found to be the most promising, robust and recyclable (3 times) catalyst that gave allylation products in 52–90% yield and 65–99% ee with various substituted aldehydes at 0 °C.Results and discussion
Recently we have shown that tosylated phenylalanine-based amides are efficient organocatalysts for the enantioselective allylation of aldehydes. Therefore, instinctively we thought of combining tosylated phenylalanine and oxazoline and synthesising organocatalyst (S,S)-1 in three simple synthetic steps. The same strategy was used to prepare the remaining organocatalysts 1–5 with varied chirality (Scheme 1).15,17 We also synthesized catalyst (S,S)-6 (Scheme 2) by replacing the tosyl part of the catalyst (S,S)-4 with Boc to ascertain the role of the tosyl group in the allylation reaction. | ||
| Scheme 1 Synthesis of tosyl-protected organocatalysts. | ||
 | ||
| Scheme 2 Synthesis of Boc-protected organocatalyst. | ||
To begin with, catalyst (S,S)-1 was evaluated for its efficacy in the asymmetric allylation of aldehydes by using 4-methoxy benzaldehyde (0.5 mmol) as a model substrate with allyltrichlorosilane (1.2 equivalent) as an allylating agent in CH2Cl2 at RT. In all of the catalytic reactions, diisopropylethyl amine (DIPEA) as a base additive (2 equiv. with respect to the substrate) was used to facilitate the allylation reaction. Catalyst (S,S)-1 showed some hopeful results (Table 1, entry 1; yield 60%, ee 54%). Catalyst (R,S)-1 with mismatched chirality gave a product with significantly lower yield (56%) and ee (40%) (entry 2).
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a All the reactions were carried out by using the substrate 4-methoxy benzaldehyde (0.5 mmol), allyltrichlorosilane (0.6 mmol), diisopropylethyl amine (1 mmol) and catalyst (10 mol%) in CH2Cl2 at RT.b Isolated yields after column chromatography.c ee determined by chiral HPLC using a Daicel Chiralcel OD-H column.d Absolute configurations were assigned by comparing both the retention time and optical rotation with the reported reliable data. | |||||||||||
 |
 |
 |
|||||||||
| Entry | Yieldb [%] | eec [%] | Config.d | Entry | Yieldb [%] | eec [%] | Config.d | Entry | Yieldb [%] | eec [%] | Config.d |
| 1 | 60 | 54 | S | 2 | 56 | 40 | S | 3 | 55 | 40 | R |
 |
 |
 |
|||||||||
| Entry | Yieldb [%] | eec [%] | Config.d | Entry | Yieldb [%] | eec [%] | Config.d | Entry | Yieldb [%] | eec [%] | Config.d |
| 4 | 40 | 17 | S | 5 | 65 | 60 | S | 6 | 72 | 70 | S |
 |
 |
 |
|||||||||
| Entry | Yieldb [%] | eec [%] | Config.d | Entry | Yieldb [%] | eec [%] | Config.d | Entry | Yieldb [%] | eec [%] | Config.d |
| 7 | 60 | 20 | R | 8 | 63 | 35 | S | 9 | 55 | 45 | S |

The opposite enantiomeric catalyst (S,R)-1 gave similar activity (yield 55%) and enantioselectivity (ee 40%) but with the opposite configuration of the allylation product (entry 3).
Based on these results, it can be concluded that the enantioinduction in the product is largely governed by the chirality originating from the phenylalaninol part of the catalyst. In order to improve the efficiency of the organocatalyst, we replaced the (S)-phenylalaninol moiety from catalyst (S,S)-1 with (S)-phenylglycinol and synthesized catalyst (S,S)-2. Unfortunately, there was a significant drop in the product yield (40%) as well as the ee (17%) (entry 4). Catalyst (S,S)-3, which was prepared by replacing the (S)-phenylglycinol moiety from catalyst (S,S)-2 with (S)-tert-leucinol, showed relatively better performance (entry 5: yield, 65%; ee, 60%). To our pleasant surprise, substitution of tosylated (S)-phenylalanine from catalyst (S,S)-3 with tosylated (S)-tert-leucine to obtain catalyst (S,S)-4 resulted in a substantial improvement in the product yield (72%) and ee (70%) (entry 6). For further improvement in the catalyst performance, we varied the steric features in the oxazoline part of the catalyst as in catalysts (S,R,S)-5 (entry 7) and (S,S,R)-5 (entry 8), but these could not match the performance of catalyst (S,S)-4. The sulfonamide moiety in organocatalyst (S,S)-4 has a specific role, possibly in activating the silicon atom of allyltrichlorosilane through coordination of its sulfonamide oxygen.
To verify this, we prepared catalyst (S,S)-6 by replacing the tosyl part of catalyst (S,S)-4 by Boc (Scheme 2), and observed a significant drop in both the product yield (55%) and ee (45%) (entry 9).
Catalyst (S,S)-4, which was the best performer, was subjected to optimization of the catalytic reaction conditions to further improve the yield and enantioselectivity of the allylation product. The role of additives in the asymmetric allylation reaction is well documented,2 therefore we first scrutinized the effect of different additives (mostly Lewis bases), viz., DIPEA, Et3N, 1,8-diazabicycloundec-7-ene (DBU) and tetrabutylammonium iodide (TBAI) (Table 2, entries 2–5), on this reaction by keeping the other parameters constant. It is to be noted that in the absence of an appropriate additive, the catalyst efficiency was significantly lower (Table 2, entry 1). DIPEA was found to be the most suitable (entry 2) among the additives used here.
|
|||||
|---|---|---|---|---|---|
| Entry | Additive [equiv.] | Temp. [°C] | Time [h] | Yieldb [%] | eec [%] |
| a Reaction conditions as per Table 1.b Isolated yield after column chromatography.c ee determined by chiral HPLC using a Daicel Chiralcel OD-H column. | |||||
| 1 | — | RT | 24 | 40 | 58 |
| 2 | DIPEA (2.0) | RT | 20 | 72 | 70 |
| 3 | Et3N (2.0) | RT | 20 | 77 | 50 |
| 4 | DBU (2.0) | RT | 20 | 68 | 41 |
| 5 | TBAI (2.0) | RT | 20 | 50 | 30 |
| 6 | DIPEA (1.0) | RT | 20 | 55 | 68 |
| 7 | DIPEA (3.0) | RT | 20 | 71 | 70 |
| 8 | DIPEA (2.0) | 0 | 24 | 67 | 97 |
| 9 | DIPEA (2.0) | −20 | 24 | 62 | 96 |
| 10 | DIPEA (2.0) | −40 | 36 | 60 | 90 |

Even the amount of DIPEA with respect to the substrate amount was found to be crucial for the desired outcome of the reaction, as is evident from the experiments carried out over a range of 1–3 equivalents of DIPEA (entries 2, 6 and 7), with 2 equivalents (entry 2) found to be the optimum. The temperature effect (Table 2, entries 8–10) on this reaction, studied from −40 °C to RT, revealed that at 0 °C the product ee (97%) was highest with 67% yield in 24 h (entry 8).
Further, the catalyst loading of 10 mol%, which was used in the foregoing experiments, was found to be optimum as it was observed that by decreasing the catalyst loading (5 mol%) the product yield (50%) dropped significantly. On the other hand, with an increase in catalyst loading (15 mol%), the reaction was faster but with a marginal drop in the ee (90%) while the product yield remained similar (Table 3, entry 3). Next, solvent variation studies (Table 3, entries 2, 4–7) showed that CH2Cl2 was the most suitable solvent among the solvents studied here (Table 3, entry 2).
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. loading [mol%] | Solvent | Time [h] | Yieldb [%] | eec [%] |
| a Reaction conditions as per Table 1.b Isolated yield after column chromatography.c ee determined by chiral HPLC using a Daicel Chiralcel OD-H column. | |||||
| 1 | 5 | CH2Cl2 | 24 | 50 | 96 |
| 2 | 10 | CH2Cl2 | 24 | 67 | 97 |
| 3 | 15 | CH2Cl2 | 17 | 68 | 90 |
| 4 | 10 | THF | 30 | 52 | 67 |
| 5 | 10 | CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (1:1) :THF (1:1) |
24 | 55 | 60 |
| 6 | 10 | CHCl3 | 20 | 60 | 70 |
| 7 | 10 | Toluene | 36 | 40 | 55 |

After achieving the optimum reaction conditions (Table 3, entry 2) for the enantioselective allylation reaction, we investigated the efficacy of the catalyst (S,S)-4 for various aromatic, hetero-aromatic and aliphatic aldehydes as substrates (Table 4).
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Time [h] | Yieldb[%] | eec [%] | Config.d |
| a Reaction conditions as per Table 1.b Isolated yields after column chromatography.c ee determined by chiral HPLC using Daicel Chiralcel and Chiralpak OD-H, AS-H, IA, IB and IC columns according to reported procedures.d Absolute configurations were assigned by comparing both the retention time and optical rotation with the reported reliable data.e Optical rotation for product 2c was negative. | |||||
| 1 | C6H5CHO (1a) | 18 | 75 | 93 | S |
| 2 | 4-MeO–C6H4CHO (1b) | 24 | 67 | 97 | S |
| 3 | 4-(C6H5CH2O)–C6H4CHO (1c) | 24 | 68 | >99 | (−)e |
| 4 | 4-(CH3)3C–C6H4CHO (1d) | 24 | 70 | 97 | S |
| 5 | 4-Me–C6H4CHO (1e) | 24 | 65 | 86 | S |
| 6 | 4-NO2–C 6H4CHO (1f) | 20 | 70 | 90 | S |
| 7 | 4-F–C6H4CHO (1g) | 22 | 78 | 75 | S |
| 8 | 4-CF3–C6H4CHO (1h) | 20 | 75 | 77 | S |
| 9 | 3-MeO–C6H4CHO (1i) | 20 | 77 | 93 | S |
| 10 | 2-MeO–C6H4CHO (1j) | 20 | 84 | 64 | S |
| 11 | 2-F–C6H4CHO (1k) | 22 | 74 | 69 | S |
| 12 | 2-Naphthaldehyde (1l) | 14 | 85 | 81 | S |
| 13 | Thiophene-2-carbaldehyde (1m) | 14 | 82 | 92 | S |
| 14 | (E)C6H5CHCHCHO (1n) | 15 | 77 | 80 | R |
| 15 | (E)C6H5CHC(CH3)CHO (1o) | 15 | 90 | 93 | R |
| 16 | C6H5CH2CHO (1p) | 24 | 52 | 67 | R |

Among the different aromatic aldehydes, m/p-substituted benzaldehydes (Table 4, entries 2–9) gave better ees as compared to their o-counterparts (Table 4, entries 10 and 11). The enantioselectivity was comparatively better for substrates with electron donating substituents (Table 4, entries 2–4) than electron withdrawing substituents (Table 4, entries 6–8). The bulkier aldehydes such as 2-naphthaldehyde, were found to be more reactive in the present catalytic system, which gave a product with higher yield, but with a significantly lower ee (Table 4, entry 12) than benzaldehyde (Table 4, entry 1). The heteroaromatic aldehyde ,thiophene-2-carboxaldehyde (Table 4, entry 13) behaved just like benzaldehyde by giving only a marginally higher yield (82%) and comparable ee (92%). α,β-Unsaturated aldehydes, viz. trans-cinnamaldehyde and trans-α-methyl cinnamaldehyde, as substrates gave products with good yields and good to excellent ees (Table 4, entries 14 and 15), but with an inverted configuration (R) as compared to the products (configuration S) obtained with the rest of the aldehydes used in the present study. Similarly, the inversion of configuration was also observed for the product (configuration R) obtained with phenylacetaldehyde, but with a significantly lower yield (52%) and ee (67%).
Reaction mechanism: NMR experiments
A series of NMR experiments were performed for understanding of the mechanism of the allylation reaction operating in the present catalytic system. For this, we looked for any observable changes in the NMR spectra of catalyst (S,S)-4 after the sequential addition of allyltrichlorosilane, substrate (benzaldehyde) and DIPEA. The 13C NMR spectra (Fig. 1) showed that the imine carbon peak (163.82 ppm) of catalyst (S,S)-4 was shifted downfield to 179.02 ppm upon the addition of allyltrichlorosilane. This large shifting (15.2 ppm) indicates that there is a strong interaction between the oxazoline nitrogen and silicon of allyltrichlorosilane to form an intermediate I-1 (Scheme 3).4l,7c | ||
| Fig. 1 13C NMR spectra were taken in CDCl3: (a) benzaldehyde; (b) catalyst (S,S)-4; (c) 13C spectrum of catalyst (S,S)-4 after interaction with allyltrichlorosilane; (d) 13C spectrum of catalyst (S,S)-4 after interaction with allyltrichlorosilane and benzaldehyde; (e) 13C spectrum of catalyst (S,S)-4 after interaction with allyltrichlorosilane, benzaldehyde and DIPEA. | ||
 | ||
| Scheme 3 Proposed catalytic cycle for the enantioselective allylation reaction catalysed by (S,S)-4. | ||
After the addition of the substrate, this peak (179.02 ppm) only marginally shifted to appear at 178.93 ppm (spectrum (d), Fig. 1). It is to be noted that on adding DIPEA, product formation takes place (full spectrum in ESI†) with concomitant release of the catalyst7c,15 (Scheme 3), as evident from spectrum (e) (Fig. 1), where the oxazoline imine carbon peak of the free catalyst at 163.42 ppm was restored (full spectrum in ESI†).
The product formation in the above reaction was followed by 1H NMR experiments. The reaction was conducted in CDCl3 with 10 mol% of catalyst (S,S)-4 for the allylation of benzaldehyde at RT, and 1H NMR spectra were recorded at 0, 4, 8 and 12 h intervals (Fig. 2). The allylation product peaks at 5.05–5.08 ppm grew over a period of time and became sufficiently visible at ∼60% conversion (full spectra in ESI†).
 | ||
| Fig. 2 1H NMR spectra were recorded in CDCl3 following the general allylation reaction procedure as described in the experimental section, with 0.5 mmol substrate (benzaldehyde), 0.6 mmol allyltrichlorosilane, 1 mmol DIPEA and 10 mol% (S,S)-4 organocatalyst. | ||
A catalytic cycle (Scheme 3) based on the results obtained during the catalyst screening experiments (Table 1) and NMR study has been proposed here. Accordingly, the catalyst first interacts with allyltrichlorosilane to form an intermediate I-1.4l,7c The intermediate I-1 then reacts with the substrate and probably forms intermediate I-2,4l,7c which reacts with DIPEA to give the desired product and releases the catalyst for the next cycle.
Recyclability of the catalyst
The main problem with homogeneous catalysts is their separation and reuse from the reaction medium in post-catalysis workup. Nevertheless, we have attempted the recycling of catalyst (S,S)-4 in order to make the present catalytic protocol more cost-effective and also to demonstrate the robust nature of the catalyst under the allylation reaction conditions used. The reuse experiments were conducted using 4-methoxy benzaldehyde as a representative substrate with catalyst (S,S)-4 and allyltrichlorosilane as the allyl source at 0 °C in CH2Cl2 in the presence of DIPEA. After the first catalytic run, the amount of solvent was reduced, and the organocatalyst (S,S)-4 was precipitated by the addition of an excess amount of hexane. The precipitate contained both organocatalyst (S,S)-4 and molecular sieves. It was then thoroughly washed with hexane and dissolved in CH2Cl2 and immediately filtered. The filtrate was then evaporated under vacuum and dried under an inert atmosphere. It was then used for the subsequent catalytic run without further purification. The recovered catalyst worked well for the three cycles studied (Fig. 3), with only a marginal loss in yield, which is attributed to the physical loss of the catalyst during the recovery process. Moreover, the 1H NMR spectra of the isolated catalyst (see ESI†) showed no change or additional peaks as compared to the fresh catalyst. | ||
| Fig. 3 Recyclability of catalyst (S,S)-4 using 4-methoxy benzaldehyde as the representative substrate. | ||
Conclusions
A series of oxazoline-based chiral organocatalysts were developed, among which the catalyst (S,S)-4 showed very good catalytic efficiency in the asymmetric allylation reaction of a wide range of substrates such as aromatic, hetero-aromatic and α,β-unsaturated aldehydes, with allyltrichlorosilane as the allyl source, under mild reaction conditions. The aromatic and hetero-aromatic aldehydes, in particular, gave allylation products with good yield (up to 90%) and excellent ee (up to 99%). The catalyst was found to be robust and showed excellent recyclability. Also, the mechanism of the asymmetric allylation reaction was proposed based on the experimental observations and evidences obtained from the NMR studies.Experimental section
The different aldehydes and reagents were used as received. All of the solvents were dried using standard procedures,19 and were distilled and stored under activated molecular sieves. NMR spectra were obtained with a Bruker F113V spectrometer (200 and 500 MHz) and were referenced internally with tetramethylsilane (TMS). Splitting patterns were reported as s, singlet; d, doublet; dd, doublet of doublets; t, triplet; q, quartet; m, multiplet; br, broad. Enantiomeric excess (ee) values were determined by HPLC (Shimadzu SCL-10AVP) using Daicel Chiralpak OD-H, AS-H, IA, IB and IC chiral columns with 2-propanol–hexane as the eluent. For the product purification, flash chromatography was performed using silica gel 100–200 mesh.General procedure for the preparation of catalysts (S,S)-1 to (S,S,R)-5
Typical procedure for preparation of catalyst (S,S)-6
General procedure for the catalytic asymmetric allylation of aldehydes with allyltrichlorosilane using (S,S)-4 as the organocatalyst
In a N2 atmosphere glove box, 18.32 mg (0.05 mmol) of catalyst (S,S)-4 was weighed out into an oven-dried 5 mL reactor. To this, 15 mg of powdered activated 4 Å molecular sieves was immediately added. Next, 1 mL of dry CH2Cl2 was added. The reactor was sealed with a septum and Teflon tape and taken out of the glove box. It was then cooled to 0 °C. To this, allyltrichlorosilane (1.2 equiv. with respect to the aldehyde) was added dropwise. After 2 h, the aldehyde (0.5 mmol) was added over 30 minutes followed by DIPEA (2 equiv. with respect to the aldehyde) was added and the reaction was allowed to stir for 24 h at this temperature. The reaction was quenched with aqueous NaHCO3 (2 mL) and extracted with dichloromethane (3 × 15 mL). The organic layers were washed with brine and dried over Na2SO4. The products were purified by flash chromatography using silica gel 100–200 mesh. The products which were confirmed by NMR data corresponded to those published previously.Acknowledgements
Authors thank CSIR for the financial support. Debashis Ghosh is thankful to AcSIR for the Ph.D. enrolment. Analytical Discipline and Centralized Instrumental Facility is gratefully acknowledged for providing instrumental facilities.Notes and references
- For selected examples of asymmetric allylation reactions used in total synthesis, see: (a) A. Fürstner and K. Langemann, J. Am. Chem. Soc., 1997, 119, 9130–9136 CrossRef; (b) D. Meng, P. Bertinato, A. Balog, D.-S. Su, T. Kamenecka, E. J. Sorensen and S. J. Danishefsky, J. Am. Chem. Soc., 1997, 119, 10073–10092 CrossRef CAS; (c) H. Nakamura, K. Nakamura and Y. Yamamoto, J. Am. Chem. Soc., 1998, 120, 4242 CrossRef CAS; (d) G. E. Keck, C. A. Wager, T. T. Wager, K. A. Savin, J. A. Covel, M. D. McLaws, D. Krishnamurthy and V. J. Cee, Angew. Chem., Int. Ed., 2001, 113, 237–240 (Angew. Chem., Int. Ed., 2001, 40, 231–234) CrossRef; (e) R. A. Fernandes, A. Stimac and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 14133 CrossRef CAS PubMed; (f) S. Kobayashi, C. Ogawa, H. Konishi and M. Sugiura, J. Am. Chem. Soc., 2003, 125, 6610–6611 CrossRef CAS PubMed; (g) A. B. Smith III, W. Zhu, S. Shirakami, C. Sfouggatakis, V. A. Doughty, C. S. Bennett and Y. Sakamoto, Org. Lett., 2003, 5, 761–764 CrossRef PubMed; (h) P. A. Evans, J. Cui and S. J. Gharpure, Org. Lett., 2003, 5, 3883–3885 CrossRef CAS PubMed; (i) T. Vilaivan, C. Winotapan, V. Banphavichit, T. Shinada and Y. Ohfune, J. Org. Chem., 2005, 70, 3464–3471 CrossRef CAS PubMed; (j) G. E. Keck and A. P. Truong, Org. Lett., 2005, 7, 2153–2156 CrossRef CAS PubMed; (k) S. E. Denmark and J. Fu, Org. Lett., 2002, 4, 1951–1953 CrossRef CAS PubMed; (l) T. Itoh, M. Miyazaki, H. Fukuoka, K. Nagata and A. Ohs, Org. Lett., 2006, 8, 1295–1297 CrossRef CAS PubMed; (m) T. R. Wu and J. M. Chong, J. Am. Chem. Soc., 2006, 128, 9646–9647 CrossRef CAS PubMed; (n) S. Lou, P. N. Moquist and S. E. Schaus, J. Am. Chem. Soc., 2007, 129, 15398–15404 CrossRef CAS PubMed; (o) S. E. Denmark, C. S. Regens and T. Kobayashi, J. Am. Chem. Soc., 2007, 129, 2774–2776 CrossRef CAS PubMed; (p) K. Maki, R. Motoki, K. Fujii, M. Kanai, T. Kobayashi, S. Tamura and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 17111–17117 CrossRef CAS PubMed; (q) V. Rauniyar and D. G. Hall, J. Org. Chem., 2009, 74, 4236–4241 CrossRef CAS PubMed; (r) S. B. Han, A. Hassan, I. S. Kim and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 15559–15561 CrossRef CAS PubMed.
- (a) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2794 CrossRef CAS PubMed; (b) J. W. J. Kennedy and D. G. Hall, Angew. Chem., Int. Ed., 2003, 42, 4732–4739 CrossRef CAS PubMed; (c) M. Yus, J. C. G. Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774–7854 CrossRef CAS PubMed.
- (a) J. A. Marshall, Chem. Rev., 1996, 96, 31–48 CrossRef CAS PubMed; (b) J. A. Marshall, in Organometallics in Synthesis, ed. M. Schlosser, J. Wiley and Sons, Chichester, U.K., 2002, p. 399 Search PubMed; (c) K. M. Waltz, J. Gavenonis and P. J. Walsh, Angew. Chem., Int. Ed., 2002, 41, 3697–3699 CrossRef CAS; (d) H. Hanawa, T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2003, 125, 1708–1709 CrossRef CAS PubMed; (e) J. G. Kim, K. M. Waltz, I. F. Garcia, D. Kwiatkowski and P. J. Walsh, J. Am. Chem. Soc., 2004, 126, 12580–12585 CrossRef CAS PubMed; (f) Y.-C. Teo, K.-T. Tan and T.-P. Loh, Chem. Commun., 2005, 1318–1320 RSC; (g) R. Takita, K. Yakura, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 13760–13761 CrossRef CAS PubMed; (h) X. Zhang, D. Chen, X. Liu and X. Feng, J. Org. Chem., 2007, 72, 5227–5233 CrossRef CAS PubMed; (i) K. Zheng, B. Qin, X. Liu and X. Feng, J. Org. Chem., 2007, 72, 8478–8483 CrossRef CAS PubMed; (j) J. Huang, J. Wang, X. Chen, Y. Wen, X. Liu and X. Feng, Adv. Synth. Catal., 2008, 350, 287–294 CrossRef CAS; (k) I. S. Kim, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6340–6341 CrossRef CAS PubMed; (l) Y. Kuang, X. Liu, L. Chang, M. Wang, L. Lin and X. Feng, Org. Lett., 2011, 13, 3814–3817 CrossRef CAS PubMed; (m) Z. Li, B. Plancq and T. Ollevier, Chem. – Eur. J., 2012, 18, 3144–3147 CrossRef CAS PubMed.
- (a) M. Nakajima, M. Saito, M. Shiro and S. Hashimoto, J. Am. Chem. Soc., 1998, 120, 6419–6420 CrossRef CAS; (b) T. Shimada, A. Kina, S. Ikeda and T. Hayashi, Org. Lett., 2002, 4, 2799–2801 CrossRef CAS PubMed; (c) S. E. Denmark and J. Fu, Chem. Commun., 2003, 167–170 RSC; (d) A. Kina, T. Shimada and T. Hayashi, Adv. Synth. Catal., 2004, 346, 1169–1174 CrossRef CAS; (e) W.-L. Wong, C.-S. Lee, H.-K. Leung and H.-L. Kwong, Org. Biomol. Chem., 2004, 2, 1967–1969 RSC; (f) H. L. Kwong, H. L. Yeung, C. T. Yeung, W. S. Lee, C. S. Lee and W. L. Wong, Coord. Chem. Rev., 2007, 251, 2188 CrossRef CAS PubMed; (g) A. V. Malkov, M. Bell, M. Orsini, D. Pernazza, A. Massa, P. Herrmann, P. Meghani and P. Kočovský, J. Org. Chem., 2003, 68, 9659–9668 CrossRef CAS PubMed; (h) A. V. Malkov, L. Dufková, L. Farrugia and P. Kočovský, Angew. Chem., Int. Ed., 2003, 42, 3674–3677 CrossRef CAS PubMed; (i) J. F. Traverse, Y. Zhao, A. H. Hoveyda and M. L. Snapper, Org. Lett., 2005, 7, 3151–3154 CrossRef CAS PubMed; (j) Q. Chai, C. Song, Z. Sun, Y. Ma, C. Ma, Y. Dai and M. B. Andrus, Tetrahedron Lett., 2006, 47, 8611–8615 CrossRef CAS PubMed; (k) A. V. Malkov, P. R. López, L. Biedermannová, L. Rulíšek, L. Dufková, M. Kotora, F. Zhu and P. Kočovský, J. Am. Chem. Soc., 2008, 130, 5341–5348 CrossRef CAS PubMed; (l) A. Kadlčíková, I. Valterová, L. Ducháčková, J. Roithová and M. Kotora, Chem. – Eur. J., 2010, 16, 9442–9445 CrossRef PubMed; (m) B. Bai, L. Shen, J. Ren and H. J. Zhu, Adv. Synth. Catal., 2012, 354, 354–358 CrossRef CAS.
- (a) K. Iseki, S. Mizuno, Y. Kuroki and Y. Kobayashi, Tetrahedron Lett., 1998, 39, 2767–2770 CrossRef CAS; (b) C. Baudequin, D. Chaturvedi and S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 2623–2629 CrossRef CAS.
- S. B. Jagtap and S. B. Tsogoeva, Chem. Commun., 2006, 4747–4749 RSC.
- (a) S. E. Denmark, J. Fu, D. M. Coe, X. Su, N. E. Pratt and B. D. Griedel, J. Org. Chem., 2006, 71, 1513–1522 CrossRef CAS PubMed; (b) M. Nakajima, S. Kotani, T. Ishizuka and S. Hashimoto, Tetrahedron Lett., 2005, 46, 157–159 CrossRef CAS PubMed; (c) S. Kotani, S. Hashimoto and M. Nakajimaa, Tetrahedron, 2007, 63, 3122–3132 CrossRef CAS PubMed.
- (a) S. E. Denmark, J. Fu and M. J. Lawler, J. Org. Chem., 2006, 71, 1523 CrossRef CAS PubMed; (b) T. Nemoto, T. Hitomi, H. Nakamura, L. Jin, K. Hatano and Y. Hamada, Tetrahedron: Asymmetry, 2007, 18, 1844 CrossRef CAS PubMed.
- I. Chataigner, U. Piarulli and C. Gennari, Tetrahedron Lett., 1999, 40, 3633–3634 CrossRef CAS.
- M. Kira, K. Sato and H. Sakurai, J. Am. Chem. Soc., 1988, 110, 4599–4602 CrossRef CAS.
- Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef CAS PubMed.
- E. Bélanger, M.-F. Pouliot, M.-A. Courtemanche and J.-F. Paquin, J. Org. Chem., 2012, 77, 317–331 CrossRef PubMed.
- R. M. Angell, A. G. M. Barrett, D. C. Braddock, S. Swallow and B. D. Vickery, Chem. Commun., 1997, 919–920 RSC.
- G. J. Rowlands and W. K. Barnes, Chem. Commun., 2003, 2712–2713 RSC.
- D. Ghosh, D. Sahu, S. Saravanan, S. H. R. Abdi, B. Ganguly, N. H. Khan, R. I. Kureshy and H. C. Bajaj, Org. Biomol. Chem., 2013, 11, 3451–3460 CAS.
- D. A. Evans, G. S. Peterson, J. S. Johnson, D. M. Barnes, K. R. Campos and K. A. Woerpel, J. Org. Chem., 1998, 63, 4541–4544 CrossRef CAS.
- A. Sadhukhan, D. Sahu, B. Ganguly, N. H. Khan, R. I. Kureshy, S. H. R. Abdi, E. Suresh and H. C. Bajaj, Chem. – Eur. J., 2013, 19, 14224–14232 CrossRef CAS PubMed.
- P. Västilä, I. M. Pastor and H. Adolfsson, J. Org. Chem., 2005, 70, 2921–2929 CrossRef PubMed.
- D. D. Perrin, W. L. F. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon, New York, 1981 Search PubMed.
- S. Saravanan, A. Sadhukhan, N. H. Khan, R. I. Kureshy, S. H. R. Abdi and H. C. Bajaj, J. Org. Chem., 2012, 77, 4375–4384 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C spectra for all new and the few known compounds, and HPLC chromatograms for the homoallyl alcohols described herein. See DOI: 10.1039/c3ra47424k |
| This journal is © The Royal Society of Chemistry 2014 |