
Epitaxial growth of successive CdSe ultrathin films and quantum dot layers on TiO2 nanorod arrays for photo-electrochemical cells
Abstract
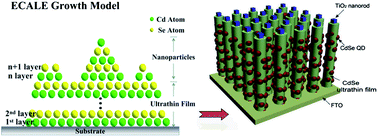
In this work, successive cadmium selenide (CdSe) ultrathin films and quantum dot layers were successfully deposited on TiO2 nanorod arrays by the electrochemical atomic layer epitaxy method (ECALE). The underpotential deposition (UPD) processes of the successive CdSe films and quantum dot layers were recorded in detail. The photo-electrochemical properties of the CdSe coated TiO2 nanorod array electrodes were also investigated, and the maximum current density reached 14.6 mA cm−2 under one sun (AM 1.5G, 100 mW cm−2). Using the ECALE method to grow a buffer layer between quantum dots and their supporting material will be useful for other energy-providing materials.
 Please wait while we load your content...
Please wait while we load your content...

