
Preparation of PDMS ultrathin films and patterned surface modification with cellulose†
Abstract
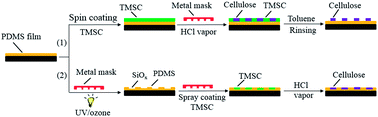
In this investigation, polydimethylsiloxane (PDMS) ultrathin films are prepared on a variety of solid surfaces by a simple and fast spin coating method, and patterned with the natural biopolymer cellulose via lithographic methods. Two surface patterning methods are developed to create coatings of hydrophilic cellulose, regenerated from trimethylsilyl cellulose (TMSC) on the PDMS thin films. In method 1, spin coated TMSC films on PDMS are covered with a lithographic mask and exposed to vapors of hydrochloric acid, which results in spatially separated cellulose pads surrounded by TMSC. Subsequent selective dissolution of TMSC with organic solvents results in a direct anchoring of cellulose pads on the PDMS. In method 2, PDMS thin films covered with a lithographic mask are exposed to UV/ozone, spray coated with TMSC and regenerated to give cellulose. The conversion of hydrophobic TMSC into hydrophilic cellulose coatings is confirmed by wettability and fluorescence measurements. The developed structures are highly transparent and stable in aqueous solutions (pH 3–9) and organic solvents. The surface properties of the polymer films are characterized using a quartz crystal microbalance with dissipation (QCM-D), ellipsometry, X-ray photoelectron spectroscopy (XPS), atomic force microscopy (AFM), contact angle and streaming potential measurements.
 Please wait while we load your content...
Please wait while we load your content...

