
Reducing disaccharides and their 1,2-dicarbonyl intermediates as building blocks for nitrogen heterocycles
Abstract
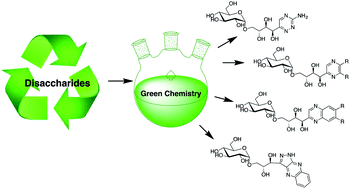
The existential importance of a sustainable economy necessitates the utilisation of plant based renewable resources such as lignin and carbohydrates. Carbohydrate utilization as industrial raw materials requires low environmental impact conversions from sugars to high value products. Here we present the conversion of reducing disaccharides into industrially relevant heterocycles of the quinoxaline-, 1,2,4-triazine-, pyrazine- and pyrazolo[3,4-b]quinoxaline-type. Heterocycle formation was facilitated by chemical conversion of reducing sugars into 1,2-dicarbonyl intermediates and their subsequent cyclization with nitrogen bis-nucleophiles. A range of disaccharides was converted into quinoxalines carrying a diverse glycosylation pattern on the polyhydroxy alkyl side chain. All transformations were performed without the need of protecting group chemistry.
 Please wait while we load your content...
Please wait while we load your content...

