First total synthesis of cryptosporiopsin A†
Barla Thirupathi and
Debendra K. Mohapatra*
Natural Products Chemistry Division, CSIR-Indian Institute of Chemical Technology, Hyderabad 500 007, India. E-mail: mohapatra@iict.res.in; dkm0077@gmail.com
First published on 10th January 2014
Abstract
The first total synthesis of polyketide natural product cryptosporiopsin A (1) was described. It has been accomplished in 12 longest linear steps with 15.4% overall yield starting from enantiomerically pure epoxide 12 prepared by hydrolytic kinetic resolution. Other key steps were Stille coupling, Grignard reaction, De Brabander's esterification and ring closing metathesis (RCM) reaction.
One of the most promising groups of natural products that have recently emerged as new lead structures for kinase inhibition is the resorcylic acid lactones (RALs), a unique class of fungal polyketide metabolites, which are β-resorcylic acid derivatives possessing a C11 side that is closed to form a 14-membered macrolactone ring (Fig. 1).1 Since the first isolation of radicicol (monorden) in 1953,2 followed by zearalenone in 1962,3 LL-Z1640-2 in 1978,4 and hypothemycin in 1980 (ref. 5) more than 30 naturally RALs have been reported. The RALs are endowed with a breadth of biological activity such as transcription factor modulations (zearalenone6 and zearalenol7), HSP90 inhibitors (radicicol8 and pochonin D9), reversible (aigialomycin D10) as well as irreversible kinase inhibitors (hypothemycin,11 IL-Z1640-2,12 and L-783277 (ref. 13)), antifungal,14 antimalarial,15 antiparasitic,16 cytotoxic,17 oestrogenic,18 nematicidal,19 protein tyrosine kinase and ATPase inhibition activities.20
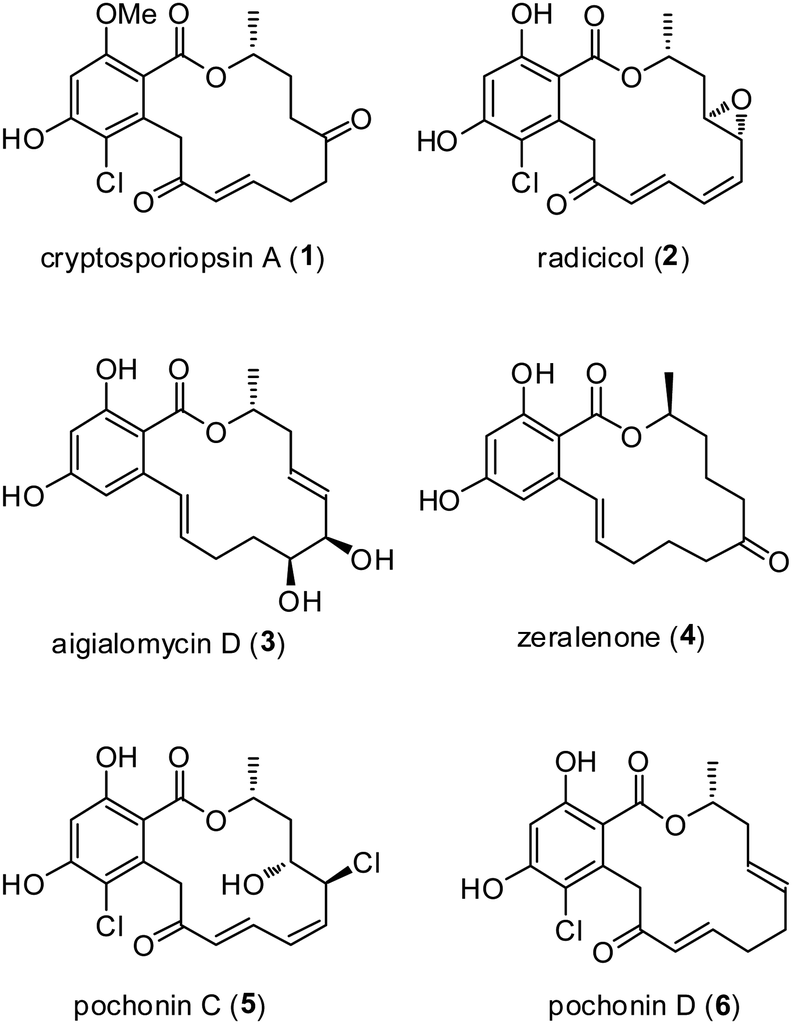 | ||
| Fig. 1 Structures of some resorcyclic acid lactones (RALs). | ||
It can thus be safe to be argued that the RAL framework is privileged21 and that even analogues of these natural products should be of interest. Recently, Laatsch et al.22 reported the isolation of a new resorcyclic acid lactone, cryptosporiopsin A (1) from Cryptosporiopsis sp. an endopyhtic fungus from leaves and branches of Zanthoxylum leprieurii (Rutaceae). The relative and absolute configuration of cryptosporiopsin A was assigned on the basis of spectroscopic and spectrometric data. Cryptosporiopsin A (1) showed motility inhibitory and lytic activities against zoospores of the grapevine downy mildew pathogen Plasmopara viticola as well as potent inhibitory activity against mycelial growth of phytopathogens, Pythium ultimum, Aphanomyces cochlioides and a basidiomycetous fungus Rhizoctonia solani. It also exhibited weak cytotoxic activity against brine shrimp larvae.22 Because of its promising biological activities and scarcity of the target natural product 1, we became interested in its total synthesis.
Construction of macrolactone through the formation of C–C bond and particularly by intramolecular ring closing metathesis (RCM) reaction stands as a promising tool for the synthesis of macrolides and heterocycles.23 As part of our ongoing program in exploring ring-closing metathesis for macrolide syntheses,24 we demonstrated the use of RCM for the total synthesis of cryptosporiopsin A. According to our retrosynthetic analysis of cryptosporiopsin A, shown in Scheme 1, could be achieved through RCM reaction of 8 which in turn could be obtained by coupling of lactone 9 and alcohol 10. Lactone 9 and alcohol 10 could be obtained from 2,4,6-trihydroxy benzoic acid 11 and epoxide 12, respectively.
 | ||
| Scheme 1 Retrosynthetic analysis of cryptosporiopsin A. | ||
Results and discussion
The synthesis of compound 10 was started from enantiomerically pure epoxide 12 in seven steps (Scheme 2). Epoxide 12 was prepared by Jacobsen's hydrolytic kinetic resolution from racemic epoxide in 45% yield with 99% ee (by HPLC).25 Treatment of the epoxide with LiAlH4 in THF furnished the corresponding secondary alcohol 13 in 85% yield.26 The hydroxyl group present in 13 was protected as its PMB ether to obtain 14 in 91% yield. Benzyl ether present in 14 was selectively deprotected27 by Raney Ni in 88% yield and the resulting primary alcohol was converted to its corresponding aldehyde by using BAIB, TEMPO28 and subsequently used for the Grignard reaction to get the desired product 16 in (with ≈1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomers by NMR and HPLC) 72% yield over two steps. The hydroxyl group present in 16 was protected as its TBS ether using TBDMSCl and imidazole in CH2Cl2 at room temperature to afford 17 (≈1:1 diastereomeric mixture) in 96% yield. PMB group present in 17 was selectively deprotected by careful treatment of DDQ29 in CH2Cl2–H2O (9:1) to produce compound 10 (≈1:1 diastereomeric mixture) in 89% yield.
:1 diastereomers by NMR and HPLC) 72% yield over two steps. The hydroxyl group present in 16 was protected as its TBS ether using TBDMSCl and imidazole in CH2Cl2 at room temperature to afford 17 (≈1:1 diastereomeric mixture) in 96% yield. PMB group present in 17 was selectively deprotected by careful treatment of DDQ29 in CH2Cl2–H2O (9:1) to produce compound 10 (≈1:1 diastereomeric mixture) in 89% yield.
 | ||
| Scheme 2 Reagents and conditions: (a) LiAlH4, THF, 0 °C–rt, 3 h, 85%; (b) PMBBr, NaH, 0 °C–rt, 8 h, 91%; (c) Raney Ni, Ethanol, 4 h, rt, 88%; (d) TEMPO, BAIB, CH2Cl2, 30 min, rt, 3-butenyl magnesium bromide, −78 °C–rt, 72% over two steps; (e) imidazole, TBSCl, CH2Cl2, 0 °C–rt, 4 h, 96%; (f) DDQ, CH2Cl2:H2O (9:1), 0 °C, 2 h, 89%. | ||
The aromatic coupling fragment was prepared in six steps starting from readily available 2,4,6-trihydroxy benzoic acid monohydrate (11) which on treatment with trifluoroacetic acid, trifluoroacetic anhydride and acetone to obtain compound 18 in 55% yield (Scheme 3).30 The hydroxyl group present in 18 was selectively protected as its PMB ether by using Mitsunobu conditions31 to afford 19 in 85% yield. Compound 19 was easily converted to its triflate with triflic anhydride in pyridine.32 Then it was subjected to allylation in presence of catalytic amount Pd(PPh3)4 in anhydrous DMF to give 20 in 76% yield over two steps.24c,33 By utilizing Jin's one-step dihydroxylation–oxidation protocol34 terminal double bond present in compound 20 was converted to corresponding aldehyde 21, which was subsequently subjected to Grignard reaction with vinyl magnesium bromide at −78 °C to furnish 22 in 87% yield. Allylic hydroxyl group present in 22 was protected as its TBS ether with TBDMSCl, imidazole to afford 9 in 92% yield.
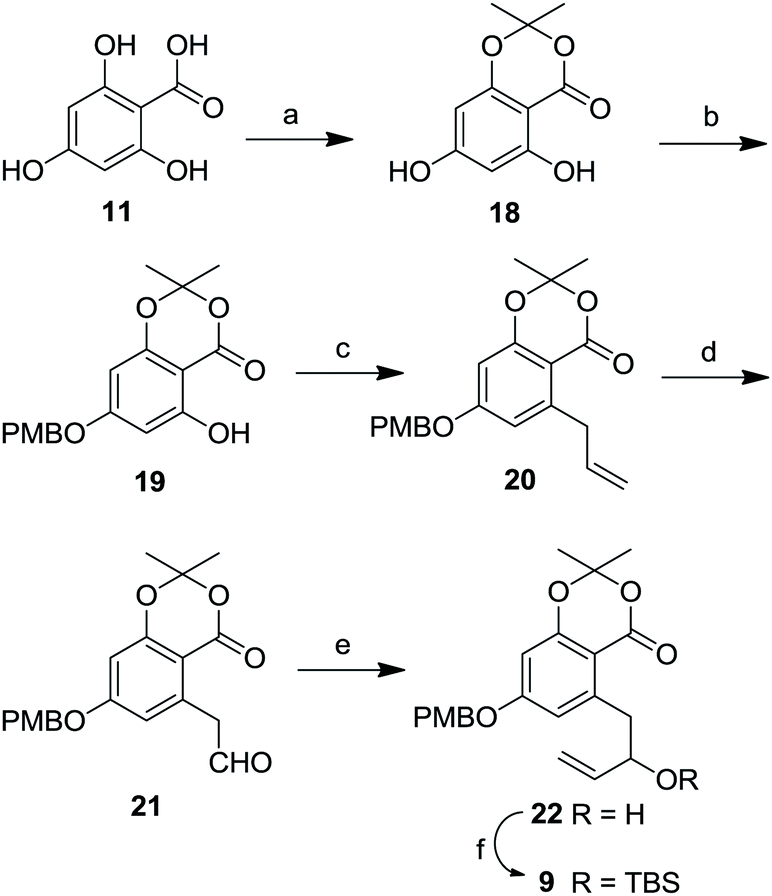 | ||
| Scheme 3 Reagents and conditions: (a) acetone, TFA, TFAA, 0 °C–rt, 12 h, 55%; (b) PMBOH, DIAD, Ph3P, CH2Cl2, 0 °C–rt, 4 h, 85%; (c) (1) Tf2O, pyridine, 0 °C–rt, 10 h; (2) Bu3Sn(allyl), LiCl, Pd(PPh3)4, DMF, 60 °C, 4 h, 76% over two steps; (d) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O, rt, 3 h, 89%; (e) vinylmagnesium bromide, THF, −78 °C, 4 h, 87%; (f) imidazole, TBDMSCl, 0 °C–rt, 3 h, 92%. | ||
After synthesizing fragments 9 and 10, coupling of these two fragments was carried out by subjecting them to De Brabanders lactonization conditions led to the formation of compound 23 (≈1:1:1:1 diastereomeric mixture) in 85% yield.35 Our next task was to introduce the chlorine functionality which was achieved by using N-chloro succinamide36 in chlorobenzene to obtain compound 8 in 77% yield (Scheme 4). As it is a mixture of four compounds, the regioselectivity of chlorination at this stage was difficult to ascertain and we thought of confirming at the later stage of the synthesis. With the required diene compound in hand which sets the stage for crucial RCM reaction and the 14-membered macrolactone formation proceeded smoothly with Grubbs second generation catalyst under refluxing conditions to afford 24 (1:1:1:1 diastereomeric mixture) in 74% yield. At this stage the geometry of the double could not be confirmed because of the presence of two racemic centers. We proceeded further to remove the silyl groups which was achieved with 1 M solution of TBAF in THF at room temperature to give 7 (≈1:1:1:1 diastereomeric mixture) in 95% yield. Oxidation of secondary alcohols to ketones was achieved by Dess–Martin periodinane in CH2Cl2 to afford compound 25 in 92% yield as a single isomer. At this stage, trans geometry of the double bond was confirmed. Treatment of the compound 25 with titanium tetrachloride (10 equiv.) in CH2Cl2 at 0 °C furnished the target molecule 1 in 87% yield. After obtaining a good NMR spectrum, we confirmed the regioselectivity of chlorination reaction by taking the help of NOE experiment. The spectroscopic and analytical properties of 1 (1H and 13C NMR, and MS) were identical to those of reported for the natural product 1. The optical rotation of synthetic 1 {[α]27D + 9.7 (c 1.05, MeOH)} was in good agreement with that of natural 1 {ref. 22 [α]20D + 11.0 (c 0.1, MeOH)}, which confirmed the absolute configuration.
 | ||
| Scheme 4 Reagents and conditions: (a) (1) NaH, 10, THF, 0 °C–rt, 5 h; (2) MeI, 0 °C–rt, 3 h, 85%; (b) NCS, chlorobenzene, 70 °C, 10 h, 77%, (c) Grubbs II, CH2Cl2, reflux, 18 h, 74%; (d) TBAF, THF, 0 °C–rt, 4 h, 95%; (e) DMP, CH2Cl2, 0 °C–rt, 5 h, 92%; (f) TiCl4, 0 °C, 10 min, 87%. | ||
Conclusions
In summary, the first total synthesis of cryptosporiopsin A (1) was accomplished in highly efficient way in 12 longest linear steps with 15.4% overall yield, starting from a known enantiomerically pure epoxide. The key steps of the synthesis were Jacobsen's hydrolytic kinetic resolution, Stille coupling, Grignard reaction, De Brabander's esterification and RCM reaction. This total synthesis has verified the absolute configuration of 1. The strategy is both concise and flexible to produce additional analogues of 1 in enantiomerically pure form. Efforts to achieve this are currently underway in our laboratory.Acknowledgements
We are thankful to the Director, CSIR-IICT and HoD, NPCD, for their kind support and encouragement. The authors thank CSIR, New Delhi for financial support as part of XII Five Year plan programme under title ORIGIN (CSC-0108). B.T. thanks the University Grant Commission (UGC), New Delhi, India for financial assistance in the form of fellowships.Notes and references
- (a) N. Winssinger and S. Barluenga, Chem. Commun., 2007, 22 RSC; (b) T. Hofmann and H. H. Altmann, C. R. Chim., 2008, 11, 1318 CrossRef CAS PubMed; (c) N. Winssinger, J. G. Fontaine and S. Barluenga, Curr. Top. Med. Chem., 2009, 9, 1419 CrossRef CAS.
- P. Delmotte and J. Delmotte-Plaquee, Nature, 1953, 171, 344 CrossRef CAS.
- M. Stob, R. S. Baldwin, J. Tuite, F. N. Andrews and K. G. Gillette, Nature, 1962, 196, 1318 CrossRef CAS.
- G. A. Ellestad, F. M. Lovell, N. A. Perkinson, R. T. Hargreaves and W. J. McGahren, J. Org. Chem., 1978, 43, 2339 CrossRef CAS.
- M. S. R. Nair and S. T. Carey, Tetrahedron Lett., 1980, 21, 2011 CrossRef CAS.
- R. J. Miksicek, J. Steroid Biochem. Mol. Biol., 1994, 49, 153 CrossRef CAS.
- W. T. Shier, Rev. Med. Vet., 1998, 149, 599 CAS.
- (a) T. W. Schulte, S. Akinaga, S. Soga, W. Sullivan, B. Stensgard, D. Toft and L. M. Neckers, Cell Stress Chaperones, 1998, 3, 100 CrossRef CAS; (b) S. V. Sharma, T. Agatsuma and H. Nakano, Oncogene, 1998, 16, 2639 CAS.
- E. Moulin, V. Zoete, S. Barluenga, M. Karplus and N. Winssinger, J. Am. Chem. Soc., 2005, 127, 6999 CrossRef CAS PubMed.
- S. Barluenga, P.-Y. Dakas, Y. Ferandi, L. Meijer and N. Winssinger, Angew. Chem., 2006, 118, 4055 CrossRef.
- (a) H. Tanaka, K. Nishida, K. Sugita and T. Yoshioka, Cancer Sci., 1999, 90, 1139 CrossRef CAS; (b) A. Schirmer, J. Kennedy, S. Murli, R. Reid and D. V. Santi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4234 CrossRef CAS PubMed.
- J. Ninomiya-Tsuji, T. Kajino, K. Ono, T. Ohtomo, M. Matsumoto, M. Shiina, M. Mihara, M. Tsuchiya and K. Matsumoto, J. Biol. Chem., 2003, 278, 18485 CrossRef CAS PubMed.
- A. Zhao, S. H. Lee, M. Mojena, R. G. Jenkins, D. R. Patrick, H. E. Huber, M. A. Goetz, O. D. Hensens, D. L. Zink, D. Vilella, A. W. Dombrowski, R. B. Lingham and L. Huang, J. Antibiot., 1999, 52, 1086 CrossRef CAS.
- (a) W. A. Ayer, S. P. Lee, A. Tsuneda and Y. Hiratsuka, Can. J. Microbiol., 1980, 26, 766 CrossRef CAS PubMed; (b) M. S. R. Nair and S. T. Carey, Tetrahedron Lett., 1980, 21, 2011 CrossRef CAS.
- V. Hellwig, A. Mayer-Bartschmid, H. Muller, G. Greif, G. Kleymann, W. Zitzmann, H.-T. Tichy and M. J. Stadler, J. Nat. Prod., 2003, 66, 829 CrossRef CAS PubMed.
- (a) M. Isaka, C. Suyarnsestakorn and M. Tanticharoen, J. Org. Chem., 2002, 67, 1561 CrossRef CAS PubMed; (b) M. Isaka, A. Yangchum, S. Intamas, K. Kocharin, E. B. G. Jones, P. Kongsaeree and S. Prabpai, Tetrahedron, 2009, 65, 4396 CrossRef CAS PubMed.
- S. Ayers, T. N. Graf, A. F. Adcock, D. J. Kroll, S. Matthew, E. J. Carcche de Blanco, Q. Shen, S. M. Swanson, M. C. Wani, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2011, 74, 1126 CrossRef CAS PubMed.
- (a) H. Boettger-Tong, L. Murthy, C. Chiappetta, J. L. Kirkland, B. Goodwin, H. Adlercreutz, G. M. Stancel and S. Makela, Environ. Health Perspect., 1998, 106, 369 CrossRef CAS; (b) B. S. Katzenellenbogen, J. A. Katzenellenbogen and D. Mordecai, Endocrinology, 1979, 105, 33 CrossRef CAS PubMed.
- J. Dong, Y. Zhu, H. Song, R. Li, H. He, H. Liu, R. Huang, Y. Zhou, L. Wang, Y. Cao and K. Zhang, J. Chem. Ecol., 2007, 33, 1115 CrossRef CAS PubMed.
- (a) E. S. Abid, Z. Ouanes, W. Hassen, I. Baudrimont, E. Creppy and H. Bacha, Toxicol. in Vitro, 2004, 18, 467 CrossRef PubMed; (b) A. Fürstner and K. Langemann, J. Am. Chem. Soc., 1997, 119, 9130 CrossRef; (c) H. J. Kwon, M. Yoshida, K. Abe, S. Horinouchi and T. Beppu, Biosci., Biotechnol., Biochem., 1992, 56, 538 CrossRef CAS; (d) P. Chanmugam, L. Feng, S. Liou, B. C. Jang, M. Boudreau, G. Yu, J. H. Lee, H. J. Kwon, T. Beppu, M. Yoshida, Y. Xia, C. B. Wilson and D. Hwang, J. Biol. Chem., 1995, 270, 5418 CrossRef CAS PubMed; (e) K. Takehara, S. Sato, T. Kobayashi and T. Maeda, Biochem. Biophys. Res. Commun., 1999, 257, 19 CrossRef; (f) N. A. Giese, and N. Lokker, Int. Pat. WO9613259.
- (a) R. Hirschmann, Angew. Chem., Int. Ed. Engl., 1991, 30, 1278 CrossRef; (b) M. A. Koch and H. Waldmann, Drug Discovery Today, 2005, 10, 471 CrossRef CAS.
- F. M. Talontsi, P. Facey, M. D. K. Tatong, M. T. Islam, H. Frauendorf, S. Draeger, A. V. Tiedemann and H. Laatsch, Phytochemistry, 2012, 83, 87 CrossRef CAS PubMed.
- (a) A. Gradillas and J. Pérez-Castells, Angew. Chem., Int. Ed., 2006, 45, 6086 CrossRef CAS PubMed; (b) A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199 CrossRef CAS PubMed; (c) R. H. Grubbs, Tetrahedron, 2004, 60, 7117 CrossRef CAS PubMed; (d) J. Prunet, Angew. Chem., Int. Ed., 2003, 42, 2826 CrossRef CAS PubMed; (e) J. A. Love, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, Germany, 2003, p. 296 Search PubMed; (f) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS PubMed; (g) A. Fürstner, Angew. Chem., Int. Ed., 2000, 39, 3012 CrossRef; (h) M. E. Maier, Angew. Chem., Int. Ed., 2000, 39, 2073 CrossRef CAS; (i) R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413 CrossRef CAS; (j) S. K. Armstrong, J. Chem. Soc., Perkin Trans. 1, 1998, 371 RSC; (k) K. Gerlach, M. Quitschalle and M. Kalesse, Tetrahedron Lett., 1999, 40, 3553 CrossRef CAS; (l) M. Nevalainen and A. M. P. Koskinen, Angew. Chem., Int. Ed., 2001, 40, 4060 CrossRef CAS.
- (a) Y. Bharat, B. Thirupathi, G. Ranjit and D. K. Mohapatra, Asian J. Org. Chem., 2013, 2, 848 CrossRef; (b) B. Jena and D. K. Mohapatra, Tetrahedron Lett., 2013, 54, 3415 CrossRef CAS PubMed; (c) B. Thirupathi, R. R. Gundapaneni and D. K. Mohapatra, Synlett, 2011, 2667 CAS; (d) D. K. Mohapatra, D. P. Reddy, U. Dash and J. S. Yadav, Tetrahedron Lett., 2011, 52, 151 CrossRef CAS PubMed; (e) D. K. Mohapatra, R. Somaiah, M. M. Rao, F. Caijo, M. Mauduit and J. S. Yadav, Synlett, 2010, 1223 CrossRef CAS PubMed; (f) D. K. Mohapatra, U. Dash, P. R. Naidu and J. S. Yadav, Synlett, 2009, 2129 CrossRef CAS PubMed; (g) D. K. Mohapatra, G. Sahoo, D. K. Ramesh and G. N. Sastry, Tetrahedron Lett., 2009, 50, 5636 CrossRef CAS PubMed; (h) D. K. Mohapatra, H. Rahman, R. Pal and M. K. Gurjar, Synlett, 2008, 1801 CrossRef CAS PubMed; (i) D. K. Mohapatra, D. K. Ramesh, M. A. Giardello, M. S. Chorghade, M. K. Gurjar and R. H. Grubbs, Tetrahedron Lett., 2007, 48, 2621 CrossRef CAS PubMed; (j) M. K. Gurjar, S. Karmakar and D. K. Mohapatra, Tetrahedron Lett., 2004, 45, 4525 CrossRef CAS PubMed; (k) M. K. Gurjar, R. Nagaprasad, C. V. Ramana, S. Karmakar and D. K. Mohapatra, ARKIVOC, 2005, 237 CrossRef.
- (a) L. P. C. Nielson, C. P. Stevenson, D. G. Blackmond and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 26, 1360 CrossRef PubMed; (b) S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307 CrossRef CAS PubMed.
- G. Sabitha, P. Padmaja, K. Sudhakar and J. S. Yadav, Tetrahedron: Asymmetry, 2009, 20, 1330 CrossRef CAS PubMed.
- I. Paterson, R. A. Ward, P. Romea and R. D. Norcross, J. Am. Chem. Soc., 1994, 116, 3623 CrossRef CAS.
- A. D. Mico, R. Maragrita, L. Parlanti, A. Vescovi and G. Piancatelli, J. Org. Chem., 1997, 62, 6974 CrossRef.
- Y. Oikawa, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1982, 23, 885 CrossRef CAS.
- R. G. Dushin and S. J. Danishefsky, J. Am. Chem. Soc., 1992, 114, 655 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS.
- L. R. Subramanian, M. Hanack, L. W. K. Chang, M. A. Imhoff, P. Schleyer, F. Effenberger, W. Kurtz, P. J. Stang and T. E. Dueber, J. Org. Chem., 1976, 41, 4099 CrossRef CAS.
- (a) F.-T. Luo and R.-T. Wang, Tetrahedron Lett., 1991, 32, 7703 CrossRef CAS; (b) S. Kamisuki, S. Takashi, Y. Mizushina, S. Anashima, K. Uramochi, S. Kobayashi, K. Sakaguchi, T. Nakata and F. Sugawara, Tetrahedron, 2004, 60, 5695 CrossRef CAS PubMed; (c) D. Martinez-Solorio, K. A. Belmore and M. P. Jennings, J. Org. Chem., 2011, 76, 3898 CrossRef CAS PubMed; (d) J. S. Yadav, N. Thrimurthulu, Md. A. Rahman, J. S. Reddy, A. R. Prasad and B. V. Subbareddy, Synlett, 2010, 3657 CAS; (e) J. S. Yadav, S. Das, J. S. Reddy, N. Thrimurthulu and A. R. Prasad, Tetrahedron Lett., 2010, 51, 4050 CrossRef CAS PubMed.
- (a) W. Yu, Y. Mei, Z. Hua and Z. Jin, Org. Lett., 2004, 6, 3217 CrossRef CAS PubMed; (b) T. K. Chakraborthy and A. K. Chattopadhyay, J. Org. Chem., 2008, 73, 3578 CrossRef PubMed; (c) T. N. Trotter, A. M. M. Alburry and M. P. Jennings, J. Org. Chem., 2012, 77, 7688 CrossRef CAS PubMed.
- A. Bhattacharjee, O. R. Sequil and J. K. De Brabander, Tetrahedron Lett., 2000, 41, 8069 CrossRef CAS.
- X. Leia and S. J. Danishefsky, Adv. Synth. Catal., 2008, 350, 1677 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and scanned copies of 1H and 13C NMR spectra for new compounds. See DOI: 10.1039/c3ra47341d |
| This journal is © The Royal Society of Chemistry 2014 |