
A highly sensitive SERS method for the determination of nitrogen oxide in air based on the signal amplification effect of nitrite catalyzing the bromate oxidization of a rhodamine 6G probe†
Abstract
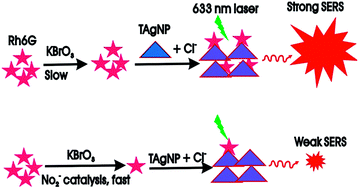
Rhodamine 6G adsorbed onto a triangular plate-type nanosilver aggregate exhibits a strong SERS peak at 1508 cm−1. Based on NO2− catalysis of the bromate oxidization of Rh6G and SERS peak quenching, trace nitrogen oxides in air samples were analyzed by a catalytic amplification SERS method.
 Please wait while we load your content...
Please wait while we load your content...

