Designed synthesis of LiNi0.5Mn1.5O4 hollow microspheres with superior electrochemical properties as high-voltage cathode materials for lithium-ion batteries
Chunyu Zhu* and
Tomohiro Akiyama*
Center for Advanced Research of Energy Conversion Materials, Hokkaido University, Sapporo 060-8628, Japan. E-mail: chunyu6zhu@gmail.com; chunyu6zhu@eng.hokudai.ac.jp; takiyama@eng.hokudai.ac.jp; Fax: +81-11-726-0731; Tel: +81-11-706-6842
First published on 31st January 2014
Abstract
LiNi0.5Mn1.5O4 hollow microspheres with designed subunits as high-voltage cathode materials for lithium-ion batteries were synthesized using dense or hollow Mn2O3 porous-spheres as self-templates. Dense Mn2O3 porous-spheres with mesosized subunits (>100 nm) were prepared by the direct decomposition of microspherical MnCO3, while the hollow and porous Mn2O3 microspheres with nanosized subunits (<30 nm) were synthesized by temperature-controlled decomposition of Mn–Ca bicarbonates followed by the selective removal of carbonates with HCl. Through a solid state reaction between Li/Ni precursors with meso/nanoporous Mn2O3, LiNi0.5Mn1.5O4 hollow spheres with different cavity size and wall structure were prepared. The as-synthesized hollow microspheres exhibited superior cyclability and high-rate capability. The best sample delivered a high and reversible discharge capacity of around 130 mA h g−1 with a capacity retention efficiency of 98.6% after 60 cycles at 1 C rate. The sample also showed high reversible capacities of 100.5 mA h g−1 even at a high current rate of 5 C. As a comparison, LiNi0.5Mn1.5O4 powders were also produced by a conventional solid state process using ball-milled Li–Ni–Mn hydroxide-oxide precursors, which showed low capacities of around 110 mA h g−1 at 1 C and greatly degraded capacities at higher current rates.
1. Introduction
The development of high-energy and high-power Li-ion batteries (LIBs) is crucial for use in mobile electronics, electric vehicles, and the storage of renewable energies, with the ultimate goals of decreasing our dependence on fossil fuels, reducing CO2 emission, and enabling clean, reliable power generation. To meet these demands, the exploration of high-voltage cathode materials plays a key role.1,2 Ni-substituted spinel material, LiNi0.5Mn1.5O4 (LNMO), is a well-known cathode material that can show excellent charge–discharge performances with a high plateau at around 4.7 V. Because of the high working voltage, the energy density of this material can reach a high value of 650 W h g−1, which can supply around 30% extra energy than those of LiMn2O4 or LiFePO4. Furthermore, the good stability, great cyclic property, low cost of raw material and environmental friendliness of this material have made it one of the most promising cathode candidates for the exploration of the next-generation LIBs.3–5Several methods have been reported to prepare LNMO, such as the solid state method,6,7 molten salt method,8 sol–gel method,9 coprecipitation method10 and others. LNMO products with various morphologies and particle sizes ranging from nanometers to microns have been synthesized by these techniques, and these products have exhibited good electrochemical performance. However, the practical use of LNMO has been constrained by many unfavorable factors, including the low-accessible capacity at high rates and poor cyclic performance due mainly to the formation of a solid-electrolyte interface (SEI) layer occurring by the undesired reaction with electrolyte, the existence of impurity phase of LixNi1−xO, and the large lattice parameter difference among the three cubic phases formed during cycling.11,12 To solve these problems, several strategies has been employed, such as the element doping,4,12,13 surface coating,14–16 and nanostructuring of the active materials to increase the intrinsic electronic conductivity by controlling the electrode structure and to enhance the Li-ion transport by reducing the mass diffusion length.17–19 Hollow structures, consisting of nanocrystallites tightly compacted to form three-dimensional channels for ion diffusion, have been reported as excellent cathode/anode materials with both high gravimetric energy density and high rate capability.17,20–23 It has been reported that hollow microspheres of spinel LiMn2O4 showed a superior rate capability and cycling stability due to the enlarged mass-electrolyte contract area, shortened Li-ion transportation distance, and enhanced electronic pathway.17,21,22
In this study, we report on the preparation of porous hollow LNMO spheres with designed nanosized subunits via a multistep synthesis: the direct decomposition of MnCO3 spheres to produce porous Mn2O3 with mesosized subunits, or a CaCO3-template method to produce porous and hollow spheres of Mn2O3 with nanosized subunits; through a solid-state reaction (SSR) between the Ni/Li precursors with the Mn2O3 precursors, LNMO hollow spheres with different cavity size and wall structure were prepared. The resulting LNMO products indicated excellent cycling performance and rate capability as compared with the sample synthesized by conventional solid state process using the ball-milled precursors.
2. Experimental
Materials preparation and characterization
The preparation for the precursors of porous Mn2O3 spheres or porous Mn2O3 hollow spheres has been described in our previous study.17 In a typical synthesis, firstly, spherical carbonates of MnCO3 and Mn3Ca1(CO3)4 were prepared using a coprecipitation method. MnSO4·5H2O, Ca(NO3)2·4H2O, and NH4HCO3 were used as starting reagents. 20 mmol of MnSO4·5H2O and Ca(NO3)2·4H2O (in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 and 3:1) were dissolved in 500 mL of distilled water, which were denoted as solution A. 100 mmol of NH4HCO3 was dissolved in 500 mL of distilled water to form solution B. 100 mL of ethanol was added to solution A under vigorous stirring. Subsequently solution B was poured into solution A and stirred to precipitate for 1 h at room temperature. The white precipitates were collected by centrifugation and washed by distilled water and ethanol for several times to remove impurities. Finally, the carbonate spheres were dried at 60 °C over night. The sample of single carbonate of MnCO3 was heated to 600 °C for 3 h to form porous Mn2O3 spheres (denoted as sample 1, S1). The coprecipitated Mn–Ca (3:1) bicarbonate was first precalcined at 400 °C for 4 h to form MnO2, which was embedded in CaCO3 with a undecomposed core. Second, the CaCO3 template and the undecomposed core were removed by washing with HCl, producing porous hollow microsphere of MnO2. Subsequently, the samples were washed by distilled water and ethanol for several times and dried at 60 °C over night. To obtain a stable manganese oxide (Mn2O3) porous hollow spheres (sample 2, S2), the dried samples were finally heat-treated at 600 °C for 3 h.
:0 and 3:1) were dissolved in 500 mL of distilled water, which were denoted as solution A. 100 mmol of NH4HCO3 was dissolved in 500 mL of distilled water to form solution B. 100 mL of ethanol was added to solution A under vigorous stirring. Subsequently solution B was poured into solution A and stirred to precipitate for 1 h at room temperature. The white precipitates were collected by centrifugation and washed by distilled water and ethanol for several times to remove impurities. Finally, the carbonate spheres were dried at 60 °C over night. The sample of single carbonate of MnCO3 was heated to 600 °C for 3 h to form porous Mn2O3 spheres (denoted as sample 1, S1). The coprecipitated Mn–Ca (3:1) bicarbonate was first precalcined at 400 °C for 4 h to form MnO2, which was embedded in CaCO3 with a undecomposed core. Second, the CaCO3 template and the undecomposed core were removed by washing with HCl, producing porous hollow microsphere of MnO2. Subsequently, the samples were washed by distilled water and ethanol for several times and dried at 60 °C over night. To obtain a stable manganese oxide (Mn2O3) porous hollow spheres (sample 2, S2), the dried samples were finally heat-treated at 600 °C for 3 h.
Cathode materials of LNMO were produced by a solid-state-reaction process using the as-prepared Mn2O3 as self-templates. The Mn2O3 porous precursors were added to a Ni(NO3)2 and LiNO3 ethanol solution in the molar ratio of Li:Ni:Mn = 1.05:0.5:1.5, followed by stirring for 2 h and drying at 80 °C in air. Finally, the dried precursors were calcined at 800 °C for 24 h to fabricate the porous hollow microspheres of LNMO. For comparison, LNMO was also produced by the conventional SSR process. The starting materials were Mn2O3, NiO, and LiOH·H2O, with the Li source in 5% excess. The starting reagents were ball-milled in a planetary-type pulverizer for 10 h in ethanol. The milled powders were dried at 100 °C to evaporate ethanol, and calcined at 800 °C for 24 h to produce LNMO.
Powder X-ray diffraction (XRD, Rigaku Miniflex, Cu-Kα) was used to analyze the phase compositions of the obtained materials. Scanning electron microscopy (SEM, JEOL, JSM-7400F) was used for the microscopic morphology observation.
Electrochemical measurement
The electrochemical characterizations were carried out in two-electrode Swagelok-type cells, with lithium metal as counter and reference electrode, 1 M LiPF6 in ethylene carbonate (EC)–dimethyl carbonate (DMC) (1:1 in volume) as electrolyte and a celgard polypropylene membrane as separator.17,24 Slurries of the LNMO active materials, conductive carbon (acetylene black), and a polyvinylidene fluoride (PVDF) binder in a weight ratio of 75:15:10, which were mixed in an N-methyl-2-pyrrolidone (NMP) solvent, were cast on an aluminum foil to form the cathodes. The cast electrodes were dried at 110 °C in a vacuum oven for 12 h and punched into 10 mm disks. The cell assembly was carried out in an Ar-filled glovebox. The cells were galvanostatically cycled between 3.3 and 4.9 V versus Li/Li+ at 25 °C using a battery tester (Arbin, MSTAT4). Cyclic voltammetry (CV) measurements were performed using a potentiostat/galvanostat apparatus (Autolab, PGSTAT128N).
3. Results and discussion
Fig. 1 shows the XRD patterns of three LNMO products. All the samples displayed the typical profile of the spinel phase (JCPDS card no. 80-2162). No obvious impurity peaks were observed. For example, the most commonly observed impurity phase of Li1−xNixO in LNMO product, with a typical peak near the LNMO (400) peak, was not detected in our products.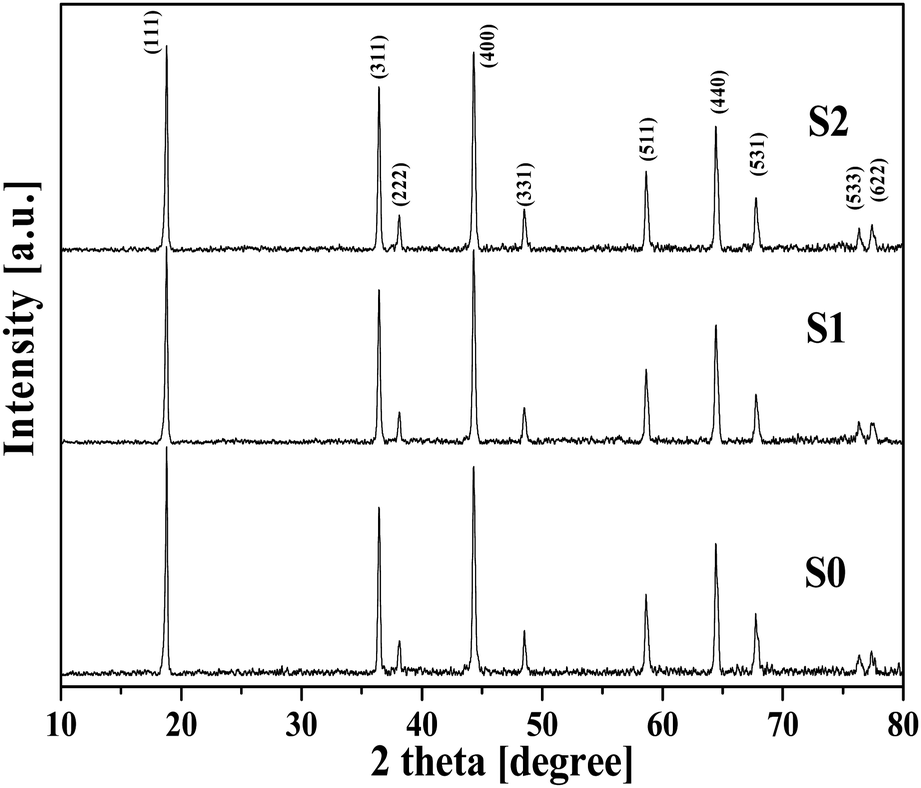 | ||
| Fig. 1 XRD patterns for the LNMO samples. | ||
The morphology and structural features of the Mn2O3 precursors and LNMO products were observed by SEM. Fig. 2(a) presents the images of the Mn2O3 precursors at different magnifications, which indicate spherical products with diameters ranging from 1 to 5 μm. The precursor of S1 was porous dense-spheres with connected primary grains larger than 100 nm, while S2 was porous hollow-spheres which consisted of nanosized subparticles as small as <30 nm. The BET specific surface areas were confirmed to be 29.45 m2 g−1 for S1 and 51.53 m2 g−1 for S2, respectively. More specification about the structural difference of these two samples was shown in our previous study.17 Fig. 2(b) shows the SEM images of the LNMO products. The sample 0 (S0), which was prepared by conventional SSR method using milled starting materials of Mn2O3, NiO, and LiOH·H2O, exhibited irregular particles in size of 200 nm to 2 μm. S1 and S2 were produced by employing the impregnation of Li and Ni reagents into the porous manganese oxide precursors followed a simple SSR process at 800 °C for 24 h. Many advantages were shown for the present synthesis route: the impregnation method provides a homogeneous distribution of the reactants on the nanoscale; the undesired grain growth during annealing is effectively suppressed for the nanosized precursors with designed voids and pores. Hollow spheres of LNMO were produced by using the porous manganese oxide spheres as precursors. The formation of hollow LNMO spheres of S1 was thought to take place because of the Kirkendall-effect during sintering although its precursor was a non-hollow porous Mn2O3.17 S1-LNMO exhibited hollow spheres with compact walls of closely agglomerated grains in the range of 100–300 nm. It is noted that almost no voids or pores were remained on the walls of the hollow S1-LNMO except for the big central holes. However, S2 showed hollow spheres with porous walls of agglomerated sub-particles in the range of 50–200 nm; the voids and pores were partially maintained for S2, and the undesired grain growth during annealing was effectively suppressed due to its loose structure and nanosized subunits (<30 nm) of the Mn2O3 hollow precursor.
 | ||
| Fig. 2 (a) SEM images for the Mn2O3 precursors and (b) SEM images for the LNMO samples at different magnification. | ||
The charge–discharge behaviour of the LNMO samples was performed in a galvanostatic mode at a potential range of 3.3 V to 4.9 V versus Li/Li+. Fig. 3(a) shows the cycling performance for the electrodes at a rate of 1 C (a rate of 1 C corresponds to a full charge–discharge of the theoretical capacity in 1 h, 1 C corresponds to 140 mA g−1 for LNMO). It is clearly observed that S2 delivered the highest reversible capacities (with discharge capacities of around 130 mA h g−1) and best capacity retention efficiency among the three samples during the 60 cycles of charge–discharge. The initial charge–discharge capacities for S0, S1, and S2 were 136.25/115.04 mA h g−1, 143.17/123.62 mA h g−1, and 152.57/131.77 mA h g−1, respectively. The three samples exhibited initial coulombic efficiencies of 84.4%, 86.3%, and 86.4%, respectively, and during cycling, the efficiencies were stabilized at around 98% for S1 and S2, which were larger than that of S0 with a value of around 97%. After 60 charge–discharge cycles, the discharge capacity retention of the three samples were 91.2%, 96.2%, and 98.6%, respectively. Fig. 3(b)–(d) show the typical galvanostatic charge–discharge curves of the electrodes at the first, 30th, and 60th cycles. It is obviously observed that S2 presented the highest stability during the cycling. However, the capacities of S0 and S1 decreased gradually and the polarization aggravated with the cycle number. The charge–discharge curves reversibly indicated two distinguishable pseudo-plateaus at around 4.70 V and a small steep plateau at 4.0 V. The former two higher plateaus were associated with the Ni2+/Ni3+ and Ni3+/Ni4+ redox reactions, while the later one was related to the Mn3+/Mn4+ redox reaction, as reported previously.18 These redox reactions were also confirmed by the CV analysis in the following section.
 | ||
| Fig. 3 Cycling performance and charge–discharge curves of the porous LNMO cathode materials at 1 C rate. | ||
To further investigate the electrochemical characteristics of the LNMO cathodes, cycling measurement were also conducted by changing the C-rates. The cells were galvanostatically charged and discharged at increasing current densities from 1 C to 5 C and then back to 1 C. Fig. 4(a) presents the discharge capacities of the LNMO electrodes at various rates. Even at a high rate of 5 C, the discharge capacity of S2 can still reached 100.5 mA h g−1, corresponding to 76.5% of its capacity at 1 C. However, for S0 and S1, the capacities decreased greatly at rates larger than 3 C, and the electrodes exhibited discharge capacities as low as around 30 mA h g−1 at a rate of 5 C. The typical charge–discharge profiles of the three electrodes at different C-rates are presented in Fig. 4(b)–(d). It is noted that the polarization at high C-rates was significantly suppressed for S2 due to its porous and hollow structure with nanosized subunits and designed voids and pores.
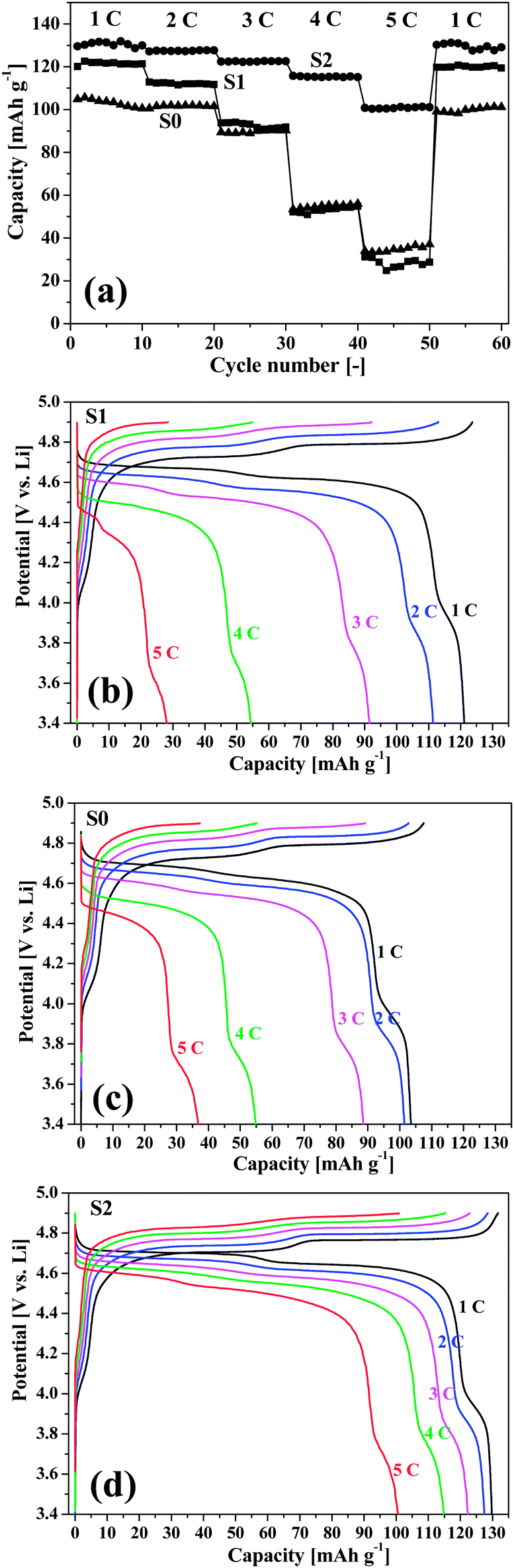 | ||
| Fig. 4 Cycling performance and charge–discharge curves of the porous LNMO cathode materials at different C rates. | ||
The LNMO hollow microspheres showed excellent cycling performance and rate capability, especially for the porous sample (S2). The enhanced properties were attributed to the unique hierarchical structure with nanosized subunits and designed voids and pores. The porous hollow structures with connected nanosized subunits facilitate rapid Li+ diffusion and efficient mass-electrolyte contract because of the significantly shortened diffusion route and a large surface-to-volume ratio, leading to a better power performance as compared to bulk electrodes. In addition, the porous hollow structure is beneficial since the connected porous framework with interior voids can effectively accommodate the strains and volume changes related to the structural transformation resulting from repeated Li+ insertion/extraction, thus retaining the capacity and improving the cycling stability.17 However, we have to keep in mind some negative side of the hollow structures, such as the decreased volumetric energy density due to their lower density with large portion of pores, when we decide the application area of the hollow cathode material with superior rate capability.
To have a further insight into the electrochemical performance, the Li intercalation/deintercalation behaviour of the LNMO electrodes were also examined by a series of cyclic voltammetry (CV) measurements. Fig. 5 shows the typical CV profiles for samples of S0, S1 and S2 at a scanning rate of 0.1 mV s−1. These curves indicated a couple of peaks at around 4.0 V and two couples of strong, separated redox peaks at 4.5–4.8 V. The strong peaks at high voltages corresponded to the redox reactions of Ni2+/Ni3+ and Ni3+/Ni4+, and the weak peaks at around 4.0 V indicated the redox couples of Mn3+/Mn4+. These were also confirmed in the charge–discharge profiles, which presented two separated plateaus at around 4.7 V and a small plateau at around 4.0 V. Obviously, the CV profile of S2 exhibited higher peak current density and more symmetrical redox peaks than that of S1 and S0, indicating the enhanced electrode reactivity.
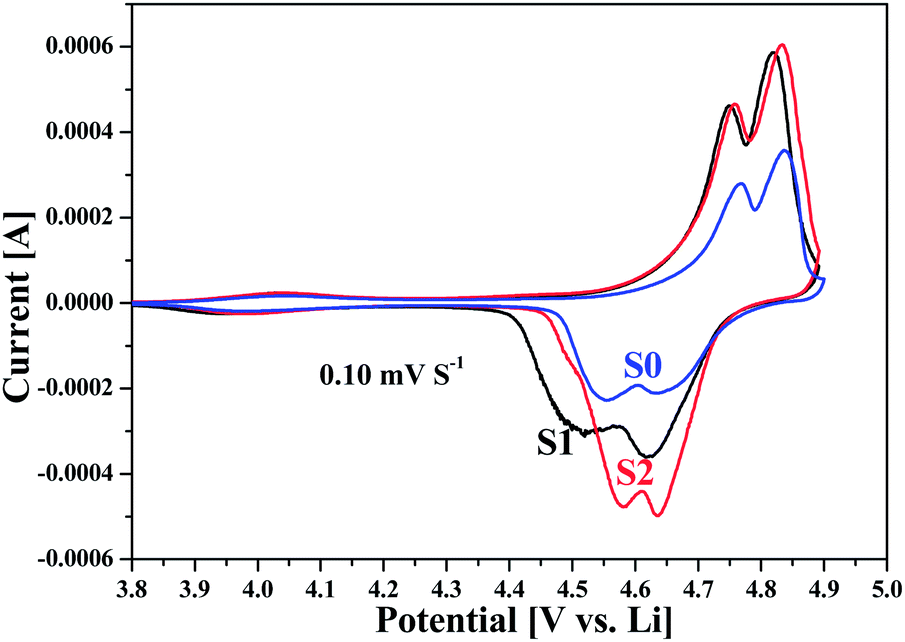 | ||
| Fig. 5 Cyclic voltammograms (CV) curves of LNMO cathode materials obtained at a scan rate of 0.1 mV S−1. | ||
4. Conclusions
In conclusion, LNMO hollow microspheres with designed subunits were synthesized using dense or hollow Mn2O3 porous-spheres as self-templates, which were reacted with the impregnated Li/Ni precursors at high temperature. LNMO hollow spheres (S1) with compact walls of closely agglomerated grains were prepared from Mn2O3 porous-spheres, which were obtained by the direct decomposition of microspherical MnCO3. LNMO hollow and porous spheres (S2) with porous walls of loosely agglomerated sub-particles (50–200 nm) and designed voids and pores were prepared from hollow and porous Mn2O3 microspheres with nanosized subunits, which were synthesized by a CaCO3-template method. S2 exhibited long-term cyclability and high-rate capability. The initial discharge capacity reached 131.77 mA h g−1 with a capacity retention efficiency of 98.6% after 60 cycles at 1 C rate. The sample also showed high reversible capacities of 100.5 mA h g−1 even at a high current rate of 5 C. However, the initial discharge capacity for S1 was only 123.62 mA h g−1 at 1 C, and at a high rate of 5 C, the capacity was as low as around 30 mA h g−1. As a comparison, LNMO powders were also produced by conventional solid state process using ball-milled Li–Ni–Mn hydroxide-oxide precursors, which showed low capacities of around 110 mA h g−1 at 1 C and greatly degraded capacities at higher current rates. Considering the remarkably improved performance, the hollow structures with designed subunits could be promising cathode materials for high-power Li-ion batteries.Acknowledgements
This work was partially supported by the Japan Society for Promotion of Science (JSPS).References
- A. Manthiram, J. Phys. Chem. Lett., 2011, 2, 176–184 CrossRef CAS.
- A. Kraytsberg and Y. Ein-Eli, Adv. Energy Mater., 2012, 2, 922–939 CrossRef CAS.
- R. Santhanam and B. Rambabu, J. Power Sources, 2010, 195, 5442–5451 CrossRef CAS PubMed.
- T.-F. Yi, Y. Xie, M.-F. Ye, L.-J. Jiang, R.-S. Zhu and Y.-R. Zhu, Ionics, 2011, 17, 383–389 CrossRef CAS.
- D. Liu, W. Zhu, J. Trottier, C. Gagnon, F. Barray, A. Guerfi, A. Mauger, H. Groult, C. M. Julien, J. B. Goodenough and K. Zaghib, RSC Adv., 2014, 4, 154–167 RSC.
- Z. Zhu, H. Yan, D. Zhang, W. Li and Q. Lu, J. Power Sources, 2013, 224, 13–19 CrossRef CAS PubMed.
- K. Mukai, Y. Ikedo, K. Kamazawa, J. H. Brewer, E. J. Ansaldo, K. H. Chow, M. Mansson and J. Sugiyama, RSC Adv., 2013, 3, 11634–11639 RSC.
- L. Wen, Q. Lu and G. Xu, Electrochim. Acta, 2006, 51, 4388–4392 CrossRef CAS PubMed.
- B. J. Hwang, Y. W. Wu, M. Venkateswarlu, M. Y. Cheng and R. Santhanam, J. Power Sources, 2009, 193, 828–833 CrossRef CAS PubMed.
- Z. Zhu, D. Zhang, H. Yan, W. Li and Qilu, J. Mater. Chem. A, 2013, 1, 5492–5496 CAS.
- T. A. Arunkumar and A. Manthiram, Electrochem. Solid-State Lett., 2005, 8, A403–A405 CrossRef CAS PubMed.
- J. Liu and A. Manthiram, J. Phys. Chem. C, 2009, 113, 15073–15079 CAS.
- E.-S. Lee and A. Manthiram, J. Mater. Chem. A, 2013, 1, 3118–3126 CAS.
- X. Fang, M. Ge, J. Rong and C. Zhou, J. Mater. Chem. A, 2013, 1, 4083–4088 CAS.
- T. Yang, N. Zhang, Y. Lang and K. Sun, Electrochim. Acta, 2011, 56, 4058–4064 CrossRef CAS PubMed.
- H. M. Wu, I. Belharouak, A. Abouimrane, Y. K. Sun and K. Amine, J. Power Sources, 2010, 195, 2909–2913 CrossRef CAS PubMed.
- C. Zhu, G. Saito and T. Akiyama, J. Mater. Chem. A, 2013, 1, 7077–7082 CAS.
- J. C. Arrebola, A. Caballero, L. Hernán and J. Morales, Eur. J. Inorg. Chem., 2008, 3295–3302 CrossRef CAS.
- J. C. Arrebola, A. Caballero, M. Cruz, L. Hernán, J. Morales and E. R. Castellón, Adv. Funct. Mater., 2006, 16, 1904–1912 CrossRef CAS.
- L. Yu, H. B. Wu and X. W. Lou, Adv. Mater., 2013, 25, 2296–2300 CrossRef CAS PubMed.
- Y.-L. Ding, X.-B. Zhao, J. Xie, G.-S. Cao, T.-J. Zhu, H.-M. Yu and C.-Y. Sun, J. Mater. Chem., 2011, 21, 9475–9479 RSC.
- L. Zhou, X. Zhou, X. Huang, Z. Liu, D. Zhao, X. Yao and C. Yu, J. Mater. Chem. A, 2013, 1, 837–842 CAS.
- J. Wang, N. Yang, H. Tang, Z. Dong, Q. Jin, M. Yang, D. Kisailus, H. Zhao, Z. Tang and D. Wang, Angew. Chem., 2013, 125, 6545–6548 CrossRef.
- C. Zhu, A. Nobuta, G. Saito, I. Nakatsugawa and T. Akiyama, Adv. Powder Technol., 2013 DOI:10.1016/j.apt.2013.05.015.
| This journal is © The Royal Society of Chemistry 2014 |