Seed-assisted synthesis of Co3O4@α-Fe2O3 core–shell nanoneedle arrays for lithium-ion battery anode with high capacity†
Yongsong Luo‡
*abe,
Dezhi Kong‡abc,
Jingshan Luob,
Yanlong Wangb,
Deyang Zhanga,
Kangwen Qiua,
Chuanwei Chengc,
Chang Ming Lid and
Ting Yu*bef
aDepartment of Physics & Electronic Engineering, Xinyang Normal University, Xinyang 464000, P. R. China
bDivision of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, 637371, Singapore. E-mail: eysluo@163.com
cShanghai Key Laboratory of Special Artificial Microstructure Materials and Technology & School of Physics and Engineering, Tongji University, Shanghai 200092, P. R. China
dInstitute for Clean Energy and Advanced Materials, Southwest University, Chongqing 400700, P. R. China
eEnergy Research Institute at Nanyang Technological University (ERIAN), 639789 Singapore
fDepartment of Physics, Faculty of Science, National University of Singapore, 117542 Singapore
First published on 26th February 2014
Abstract
A novel hierarchical Co3O4@α-Fe2O3 core–shell nanoneedle array (Co3O4@α-Fe2O3 NAs) on nickel foam substrate is synthesized successfully by a stepwise, seed-assisted, hydrothermal approach. This composite nanostructure serving as an anode material for lithium-ion batteries (LIBs) is advantageous in providing large interfacial area for lithium insertion/extraction and short diffusion pathways for electronic and ionic transport. The results show that a high initial discharge capacity of 1963 mA h g−1 at 120 mA g−1 was obtained by using these hierarchical Co3O4@α-Fe2O3 NAs heterostructures as an anode, and is retained at 1045 mA h g−1 after 100 cycles, better than that of pure Co3O4 nanoneedle arrays (Co3O4 NAs) and α-Fe2O3 film grown under similar conditions, indicating a positive synergistic effect of the material and structural hybridization on the enhancement of the electrochemical properties. The fabrication strategy presented here is facile, cost-effective, and scalable, which opens new avenues for the design of optimal composite electrode materials with improved performance.
1. Introduction
Rechargeable lithium-ion batteries (LIBs) with high energy and power density as well as lightweight design are widely used in portable electronic devices, such as computer memory chips, micro-machines, micro-sensors, drug delivery systems and medical implant devices.1–4 However, in contrast to the fast advancements of microdevices, the reduction of battery size has not kept pace with the size reduction of electronic devices, partly due to the difference in the levels of research activity and the difficulty in the manipulation of the small area of the electrode structure. Therefore, future efforts should be made to develop smaller micro-/nano-batteries with the largest capacity per footprint area possible.3,5 Insufficient power and energy from two-dimensional (2D) microbattery configurations inspires search for the development of 3D micro-/nano-batteries using cheap and light electrode materials.6–8 Compare with 2D thin film structure, 3D structures such as branched nanowire or core–shell nanosheet arrays can potentially utilize the vertical dimension to increase the active material loading while maintaining similar Li ion-transport distances. In addition, it generally provides a larger surface area to enhance the interfacial kinetics, sufficient space to accommodate the stress relaxation and a direct pathway for electron transport.9In particular, among the transition-metal oxides, cobalt oxide (Co3O4) with high theoretical capacity (890 mA h g−1), good chemical/thermal stability, environmental benignity, and low cost synthesis is a promising candidate for Li ion batteries.10–16 In the past few years, much progress has been made in improving the performance of Co3O4 based anode materials by structural and morphological design. For instance, mesoporous or hollow Co3O4 structures are obtained by assembly of cobalt salt with molecular templates17 or hard templates,18 providing large surface area and space for accommodation of volume change during battery cycling. Co3O4 nanowire19 or nanoneedle arrays20,21 grown by hydrothermal approaches are other targets of interest, due to enhanced efficiency of ion transport through these 1D nanostructures. In addition, the direct growth of Co3O4 nanowires on a conducting substrate, such as Ti or indium-doped tin oxide (ITO),22 and the incorporation of metal oxides with graphene23 or carbon nanotubes,24 have been demonstrated to enable good electrical contact and enhanced pathways for Li+ transport kinetics.22,25
Recently, it has been proposed that the hetero-junction integration of different metal oxides between shell and core represent a unique architecture, where the high surface area of shell and efficient 1D transport through the core can substantially benefit their application in LIBs.26,27 For instance, SnO2/α-Fe2O3 hetero-structures were recently reported by a chemical vapor deposition (CVD) method28 and a hydrothermal method,29 respectively. Although hematite (α-Fe2O3) has a large theoretical capacity (1007 mA h g−1),30 the short carrier diffusion length limits its application in Li+ storage. Herein, we report a facile route to fabricate the Co3O4@α-Fe2O3 NAs for the anode material of LIBs using hydrothermal method. The direct attachment and close contact of Co3O4 NAs on the current collector (Ni foam) enables fast charge transfer pathways without of adding binders or conducting additives. Moreover, α-Fe2O3 shells provide high surface area for Li+ intercalation and structural flexibility for volume change. When used as a 3D microbattery anode, it is proved by a set of electrochemical experiments that the as-grown Co3O4@α-Fe2O3 NAs electrode exhibits good electrochemical performance in terms of capacity, cycling ability and rate capability, due to the synergistic effect between these two components in an integrated structure.
2. Experimental
Synthesis of mesoporous Co3O4 nanoneedle arrays (Co3O4 NAs) on nickel foam
The Co3O4 NAs grown on Ni foam substrates were obtained according to a modified hydrothermal method. In a typical synthesis procedure: 0.58 g of Co(NO3)2·6H2O, 0.60 g of CO(NH2)2, and 0.30 g of NH4F were dissolved thoroughly in 40 mL of distilled water under vigorous stirring for 10 min at room temperature. In the meantime, one piece of nickel foam substrate (1.5 × 3.5 cm2 in size) was carefully cleaned with concentrated HCl solution (37 wt%) in an ultrasound bath for 5 min in order to remove the surface nickel oxide layer, and then washed by deionized water and absolute ethanol for 5 min each. The aqueous solution and the Ni foam were transferred into a 50 mL Teflon-lined stainless-steel autoclave. The autoclave was sealed and maintained at 120 °C for 9 h, and then allowed to cool to ambient temperature naturally. Finally, the as-received Ni foam substrate was taken out and repeatedly rinsed several times with deionized water, then dried at 60 °C for 4 h, and followed by annealing at 400 °C in air for 4 h.Preparation of Co3O4@α-Fe2O3 core–shell nanoneedle arrays (Co3O4@α-Fe2O3 NAs) on nickel foam
First, the as-prepared Co3O4 NAs with Ni foam was immersed into the 0.1 M ethanol solution of zinc acetate (Zn(CH3COO)2) for about 15 s and then dried at 70 °C. After repeated this process for several times, it was annealed at 400 °C in the Ar atmosphere for 2 h, which led to the formation of ZnO seeds on the Co3O4 NAs. Subsequently, the ZnO-coated Co3O4 nanoneedle arrays (Co3O4@ZnO NAs) were immersed into an aqueous solution of Fe(NO3)3·9H2O (0.013 M) at room temperature for 24 h, ensuring the sufficient dissolution of ZnO nanoparticles. In this solution, Fe(NO3)3·9H2O hydrolyzed to Fe(OH)3 colloid on the surface of the Co3O4 NAs while ZnO dissolved simultaneously in the solution with acids produced by FeCl3·6H2O hydrolysis (Fe3+ + 3H2O → Fe(OH)3 + 3H+). After the immersion, the sample was rinsed with deionized water and ethanol, dried at 60 °C for 5 h, and then further annealed at 450 °C in Ar gas for 3 h. In contrast, the pure α-Fe2O3 film was obtained by using bare Ni foam as growth substrates, under the same growth conditions.Structural characterization
Scanning electron microscopy (SEM) images were obtained using a JEOL JSM 6700F microscope (Japan). The crystal structure and phase purity of the obtained products were identified by X-ray powder diffraction (XRD) using a D8 Focus (Germany, Bruker) automated X-ray diffractometer system with Cu-Kα (λ = 1.5406 Å) radiation at 40 kV and 40 mA in a 2 range from 20° to 80° at room temperature. Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) observations were carried out on a JEOL JEM-2010 instrument in bright field and on a HRTEM JEM-2010FEF instrument (operated at 200 kV). Raman spectroscopy was carried out using WITEC CRM200 Raman system equipped with a 532 nm laser source and 100× objective lens. X-ray photoelectron spectroscopy (XPS) spectra were measured on a Perkin-Elmer model PHI 5600 XPS system with a resolution of 0.3–0.5 eV from a monchromated aluminum anode X-ray source.Battery fabrication and measurement
Electrochemical measurements were carried out using two-electrode cells with lithium metal as the counter and reference electrodes. The cell assembly was performed in an Ar-filled glove box (Mbraun, Unilab, Germany) by directly using the Co3O4@α-Fe2O3 NAs on Ni foam (mCo3O4 + α-Fe2O3 ≈ 6.0 mg) as the anode materials, Li metal circular foil (about 0.59 mm thick, 14 mm diameter) was used as the counter and reference electrodes, a microporous polypropylene membrane as the separator, LiPF6 (1 M) in ethylene carbonate (EC) and diethyl carbonate (DEC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume) as the electrolyte. The cell was aged for 15 h before measurement. Cyclic voltammetry was measured on a CHI660D electrochemical workstation, in the potential range of 0.01–3.0 V at a scanning rate of 0.5 mV s−1. The charge–discharge cycling was performed at room temperature by using a multi-channel battery workstation (Neware Co., China). The electrochemical impedance spectroscopy (EIS) of the electrodes was performed on a CHI660D electrochemical workstation with an ac signal voltage of 5 mV in amplitude and the frequency ranged from 100 kHz to 0.01 Hz at open circuit potential. Before the EIS measurement, the electrodes were cycled for five cycles, then discharged to 3.0 V and kept until the open circuit voltage stabilized.
:1 by volume) as the electrolyte. The cell was aged for 15 h before measurement. Cyclic voltammetry was measured on a CHI660D electrochemical workstation, in the potential range of 0.01–3.0 V at a scanning rate of 0.5 mV s−1. The charge–discharge cycling was performed at room temperature by using a multi-channel battery workstation (Neware Co., China). The electrochemical impedance spectroscopy (EIS) of the electrodes was performed on a CHI660D electrochemical workstation with an ac signal voltage of 5 mV in amplitude and the frequency ranged from 100 kHz to 0.01 Hz at open circuit potential. Before the EIS measurement, the electrodes were cycled for five cycles, then discharged to 3.0 V and kept until the open circuit voltage stabilized.
3. Results and discussion
The schematic illustration for the fabrication processes of highly ordered Co3O4@α-Fe2O3 NAs is shown in Fig. 1. The whole process involves three steps: first, the Co(OH)2 NAs are grown directly on a nickel foam substrate by a facile hydrothermal process, and after further thermal annealing, the Co3O4 NAs are obtained. It is noteworthy that these hydroxide nanoneedles are highly porous. They can be further converted to mesoporous oxides by thermal annealing. The SEM images of as-obtained Co3O4 NAs were shown in Fig. S1a and b.† The close observation reveals that a porous Co3O4 nanoneedle is typically with a length of about 6 μm and diameter of 50–100 nm in the middle part (Fig. S1b†). Second, the Co3O4 NAs were subjected to impregnation with zinc acetate ethanol solution and subsequent post-annealing in Ar gas, which lead to the uniform coating of a ZnO seeds layer on the nanoneedle surface. Finally, the Co3O4@ZnO NAs are immersed in an aqueous solution of Fe(NO3)3·9H2O at room temperature, a layer of Fe(OH)3 colloid was uniformly and compactly covered on the surface of Co3O4 NAs. Further annealing enables the formation of Co3O4@α-Fe2O3 NAs. This stepwise approach allows us to directly fabricate hierarchical nanostructures over large areas on conducting substrates. The optical images of the Co3O4 NAs, Co3O4@ZnO NAs, and integrated Co3O4@α-Fe2O3 NAs on current collector were shown in Fig. 2, respectively. It can be seen that the Ni foam surface completely turned into deep red after the initial growth of Co(OH)2 NAs. And then the Ni foam surface completely turned into deep black after calcination. The Co3O4/Ni substrate was observed to become light black after the deposition of ZnO, indicating the formation of ZnO layer onto the surface of Co3O4 NAs. Eventually, the formation of Co3O4@α-Fe2O3 NAs was still emerged with deep brown owing to a layer of α-Fe2O3 nanosheets covering the surface of Co3O4 NAs.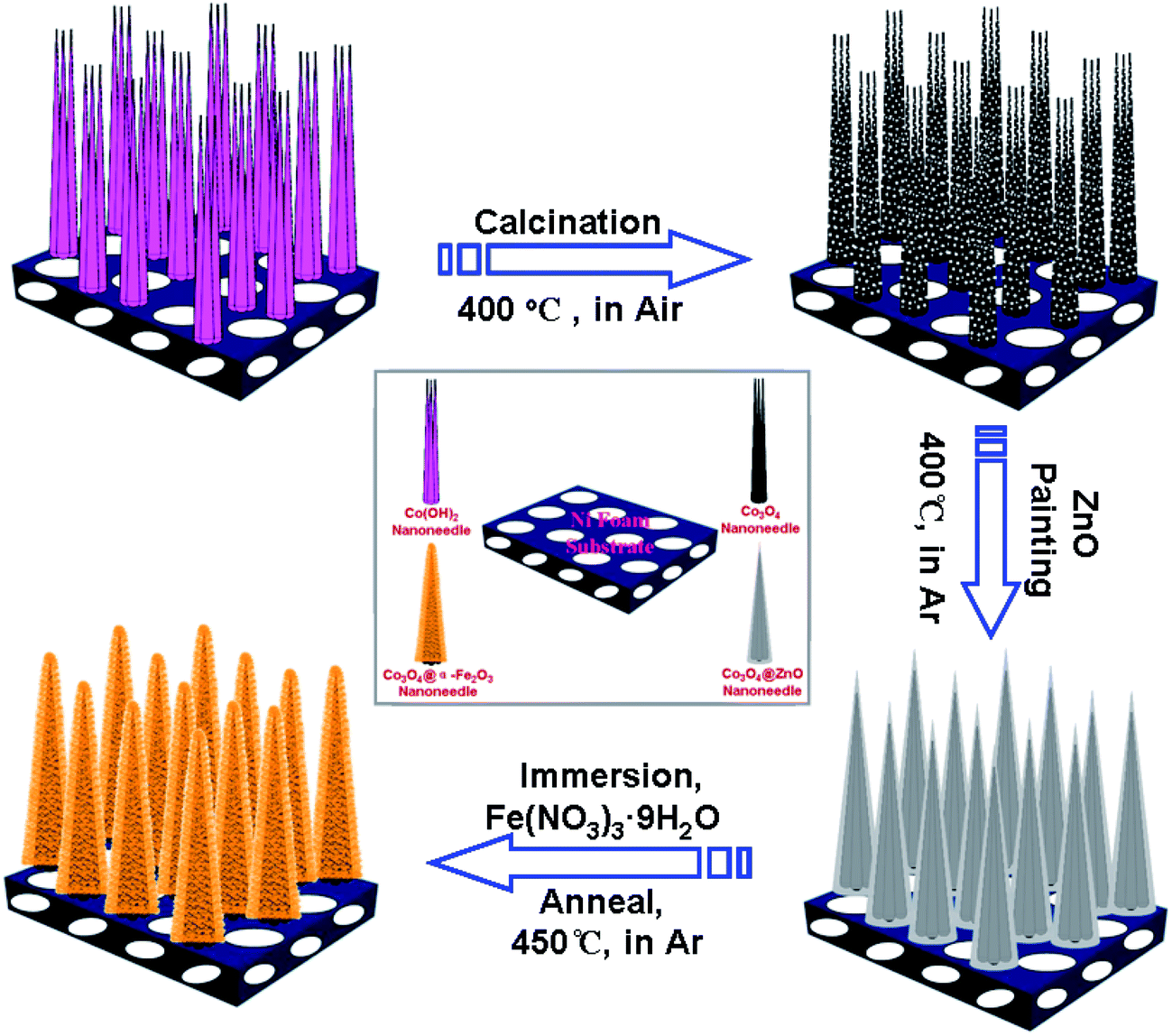 | ||
| Fig. 1 Schematic illustrating the fabrication processes of the Co3O4@α-Fe2O3 core–shell nanoneedle arrays (Co3O4@α-Fe2O3 NAs). | ||
 | ||
| Fig. 2 (a and b) Optical images of the formation processes of the Co3O4@α-Fe2O3 NAs; (c and d) low-magnification and enlarged SEM images of the pure nickel foam; (e and f) low-magnification and enlarged SEM images of the Co3O4@α-Fe2O3 NAs. | ||
The SEM images of the evolution from Co3O4 NAs to Co3O4@α-Fe2O3 NAs are shown in Fig. 3. Fig. 3a–c and g show the typical SEM images of an aligned Co3O4 nanoneedle arrays on Ni foam. It can be seen that the Co3O4 NAs are uniformly and vertically grown on the nickel foam. From a closer view in Fig. 3b and g, the Co3O4 NAs has a relatively smooth surface and a length of about 5–6 μm, and several nanoneedles gathered each other forming a bundle structure. As shown in Fig. 3d–f and h, Co3O4 NAs surface is uniformly covered with a layer of lamellar α-Fe2O3 after the α-Fe2O3 growth. This morphology is also strongly supported in Fig. S1d and S2c.† Slices of α-Fe2O3 structure of these projections can provide larger surface area and more Li+ adsorption sites, thereby enabling the core–shell composite materials with high capacitance. Fig. 3i is shown to be nanoneedle arrays of ZnO nanocrystal coating of the SEM diagram, from inserting graphics amplification can be seen in the surface of Co3O4 NAs coated ZnO nanoparticles.
 | ||
| Fig. 3 (a–c) Low-magnification and enlarged SEM images of the Co3O4 NAs; (d–f) low-magnification and enlarged SEM images of the Co3O4@α-Fe2O3 NAs; (g) cross-sectional SEM image of the Co3O4 NAs; (h) cross-sectional SEM image of the Co3O4@α-Fe2O3 NAs; (i) SEM image of the ZnO-coated Co3O4 NAs. The inset shows enlarged images. | ||
Evidence of the composition evolution is verified by X-ray diffraction (XRD). The XRD patterns of the original Co(OH)2 NAs, Co3O4 NAs, ZnO-coated Co3O4 NAs and Co3O4@α-Fe2O3 NAs are shown in Fig. 4. For curve a, almost all the diffraction peaks in this pattern can be indexed to pure hexagonal phase of β-Co(OH)2, which are in good agreement with the values in the literature (JCPDS card no. 30-0443). After annealing at 400 °C, the β-Co(OH)2 was converted into a cubic phase of Co3O4 (JCPDS card no. 42-1467; for curve b), the diffraction peaks at 19.0°, 31.3°, 36.9°, 38.5°, 59.5° and 65.3° are associated with the (111), (220), (311), (222), (511) and (440) reflections of cubic Co3O4, consistent with the previous reports.31 As for the ZnO-coated Co3O4 nanoneedle hybrids, all of the diffraction peaks can be indexed as a mixture of hexagonal wurtzite ZnO (JCPDS card no. 36-1451)32 and the tetragonal rutile Co3O4 (curve c), indicating that the Co3O4 nanoneedle array surface has a layer of ZnO. After immersing in Fe3+ solution for 24 h and further calcination in Ar at 450 °C, the ZnO peaks disappeared completely, the five new peaks located at 24.1°, 33.1°, 35.6°, 40.5° and 49.5° match well with the (012), (104), (110), (113) and (024) reflections of hexagonal α-Fe2O3 (for curve d; JCPDS card no. 33-0664),33 indicating the formation of α-Fe2O3 shells. In addition, with the exclusion of three strong peaks from the Ni foam substrate (marked with “#”), no additional impurity peaks are observed in these samples, indicating the high purity of these products.
 | ||
| Fig. 4 XRD patterns of as-grown (a) Co(OH)2 NAs, (b) Co3O4 NAs, (c) Co3O4@ZnO NAs and (d) Co3O4@α-Fe2O3 NAs. Diffraction peaks of Ni foam were marked with “#”. | ||
The surface chemical compositions and the valence states of Co3O4@α-Fe2O3 NAs were revealed by XPS (Fig. S3†). To identify all the states of cobalt, iron and oxygen element, we deconvoluted the Co 2p, Fe 2p and O 1s. The core level spectra of Co 2p were curve fitted, as shown in Fig. S3a.† Two major peaks at 779.8 and 795.2 eV are assigned to the Co 2p3/2 and Co 2p1/2, respectively. The absence of prominent shake-up satellite peaks in the Co 2p spectra further confirms the formation of the Co3O4 phase.34,35 Similarly, the core level spectra of Fe 2p were curve fitted and shown in Fig. S3b.† There are five multiple peaks for Fe 2p observed in the spectra. Peaks corresponding to 708.4 eV and 721.6 eV are attributed to +2 oxidation states, whereas 710.5 eV and 723.4 eV are ascribed to +3 states of iron. The peak centered at 715.5 eV is identified as the surface peak of α-Fe2O3.36 The deconvolution peaks (Fig. S3c†) of O 1s spectrum were also resolved into four components, which are centered at 530, 531.3, 532.7, and 534.8 eV, respectively. The low binding energy component observed at 530 eV is attributed to the O2− forming oxide with cobalt and iron elements, the later three peaks were assigned to OH−, C–O and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and H2O, respectively.37,38
O, and H2O, respectively.37,38
The structure and morphology evolution of the composite arrays were further investigated by TEM and high-resolution TEM (HRTEM). Fig. 5a shows a typical low-magnification TEM image of the tip region of Co3O4 NAs scraped off from Ni foam, confirming their needle-like nanostructures. The diameter of the tips is several nanometers. Fig. 5b is a TEM image of the middle region, showing that Co3O4 NAs are of porous structure with a rough surface and their diameter is several tens of nanometers. The fringe spacing is determined to be about 2.4 Å according to the HRTEM image in Fig. 5c, which corresponds to the (311) planar spacing of Co3O4 (JCPDS card no. 42-1467).39 In addition, the selected area electron diffraction (SAED) pattern of the Co3O4 nanoneedle demonstrates that the oxide is crystalline in nature (as shown in set of Fig. 5c). The TEM image (Fig. 5d) clearly demonstrates that most of the α-Fe2O3 nanoparticles were uniformly deposited on the surface of the Co3O4 nanoneedles and the structure of the nanoneedles was well maintained. Fig. 5e shows the magnified TEM image of an individual Co3O4@α-Fe2O3 core–shell nanoneedle. It is clear that the continuous α-Fe2O3 layers consisted of α-Fe2O3 nanosheets with a size less than 50 nm and were uniformly coated on the surface of Co3O4 nanoneedles, resulting in the formation of hybrid core–shell structures. Clear lattice fringes are observed in Fig. 5f, the fringe spacing of 0.23 nm matches well with the interplanar spacing of the (006) planes of the α-Fe2O3,2 which further demonstrates that Co3O4@α-Fe2O3 NAs has a single phase orthorhombic structure and good crystallinity.
 | ||
| Fig. 5 (a–c) Low-magnification and high-magnification TEM images of the Co3O4 NAs; (d–f) low-magnification and high-magnification TEM images of the Co3O4@α-Fe2O3 NAs. The insets of (c) and (f) are the corresponding SAED patterns from Co3O4 NAs and Co3O4@α-Fe2O3 NAs. | ||
In order to clearly reveal the evolution process of composite materials at different growth stages, we measured the Raman scattering of the Co3O4 NAs, Co3O4@ZnO NAs and Co3O4@α-Fe2O3 NAs (Fig. 6). As shown in Fig. 6a, the Co3O4 NAs exhibits four Raman active peaks at 478.6 (Eg), 519.2 (F2g), 616.6 (F2g) and 684.5 cm−1 (A1g).40 For the Co3O4 spinel, Raman mode at 684.5 cm−1 (A1g) is attributed to characteristics of the octahedral sites and the Eg and F2g modes are likely related to the combined vibrations of tetrahedral site and octahedral oxygen motions.41 After coating the ZnO seed layer on the surface of the Co3O4 NAs, in addition to the three weak peaks from Co3O4, the other five strong peaks were obtained, which are corresponding to the ZnO peak position. In the Raman spectrum of Co3O4@α-Fe2O3 NAs (as shown in Fig. 6c), the absence of ZnO Raman modes indicates the complete consumption of the ZnO nanoparticles. Meanwhile, the peaks at 225 and 498 cm−1 were assigned to A1g modes. The peaks at 247, 293, 412, and 613 cm−1 were also assigned to Eg modes. The intense peak at 1320 cm−1 is assigned to a two-magnon scattering which arise from the interaction of two magnons created on antiparallel close spin sites.42
 | ||
| Fig. 6 Raman spectra of the (a) pure Co3O4 NAs, (b) Co3O4@ZnO NAs and (c) Co3O4@α-Fe2O3 NAs, respectively. | ||
In virtue of the robust mechanical adhesion and good electrical contact enabled by the direct growth of metal oxide nanostructures on the current collector, the Co3O4@α-Fe2O3 composite nanoneedle arrays was further tested as a lithium ion battery anode without adding binders or conducting additives. The electrochemical experiments were conducted in a two-electrode configuration with the as-prepared array on the Ni foam substrate used directly as the working electrode and Li foil as the counter electrode. The electrochemical reaction mechanism of Li with Co3O4 and α-Fe2O3 has been well studied and can be expressed in the following equations:
| Co3O4 + 8Li+ + 8e− → 4Li2O + 3Co | (1) |
| Co + Li2O ↔ CoO + 2Li+ + 2e− | (2) |
| Fe2O3 + 2Li+ + 2e− → Li2(Fe2O3) | (3) |
| Li2(Fe2O3) + 4Li+ + 4e− → 2Fe0 + 3Li2O | (4) |
| 2Fe0 + 2xLi2O ↔ 2FeOx + 4xLi+ + 4xe− | (5) |
Galvanostatic measurement of discharge–charge cycles were performed at a voltage window of 5 mV to 3.0 V (vs. Li), and several representative cycles are displayed (Fig. 7a and b). At a current density of 120 mA g−1, the Co3O4@α-Fe2O3 NAs anode reaches a high initial discharge capacity of ∼1963 mA h g−1, substantially better than those of Co3O4 NAs (1338.5 mA h g−1) and α-Fe2O3 film (347.5 mA h g−1), due to the synergistic effect between the nanostructured Co3O4 and α-Fe2O3. The substantially low capacity of α-Fe2O3 nanosheets anode is ascribed to the low electrical and ionic conductivity of hematite.43 At the second cycles, the discharge capacities of all anode material decrease, corresponding to an irreversible capacity loss due to the electrolyte decomposition on the electrode surface and the SEI layer formation.44 The reversible capacity of the Co3O4@α-Fe2O3 NAs anode relatively reduces since the second cycle, however, it is retained at 1045 mA h g−1 after 100 cycles (Fig. 7d). The coulombic efficiency of each cycle is over 100%. This capacity value corresponds to 53.2% of its initial discharge capacity, and is much higher than those of pristine Co3O4 NAs (674 mA h g−1) and α-Fe2O3 nanosheets (298 mA h g−1). Notably, this reversible capacity is even higher than the theoretical capacity of Co3O4 (890 mA h g−1) and close to that of α-Fe2O3 (1007 mA h g−1),26 indicating substantial enhancement of Li+ storage capacity and stability for Co3O4@α-Fe2O3 NAs. The improved performance might be attributed to synergistic effect between Co3O4 and α-Fe2O3 and 3D structural design. The primary Co3O4 NAs trunk can serve as a one-dimensional conduction channels for fast Li+ transport, while the secondary α-Fe2O3 nanosheets provide high surface area for enhanced electrolyte accessibility as well as large electrochemical capacity for Li+ storage. In addition, the space between α-Fe2O3 shells can reduce the aggregation of primary Co3O4 NAs and alleviate the mechanical stress of volume change induced by Li+ intercalation/extraction. After long-term cycling, the morphologies of the Co3O4@α-Fe2O3 NAs are relatively well retained, although negligible collapse and amalgamation of nanoneedles are also observed (as illustrated in Fig. S1e and f†). Together with the electrochemical measurement, these results confirm our rational design of integrating active α-Fe2O3 nanosheets into the space of neighboring Co3O4 NAs, while also providing structural spacers and efficient electrolyte penetration.
 | ||
| Fig. 7 Galvanostatic discharge–charge profiles of the Co3O4@α-Fe2O3 NAs anode (a) and the Co3O4 NAs anode (b) at a constant current density of 120 mA g−1; (c) CV curves of a Co3O4@α-Fe2O3 NAs anode at 0.5 mV s−1 scanning rate; (d) cycling performance of the anodes at a constant current density of 120 mA g−1; (e) reversible capacity vs. current density (rate capability) for different anodes; (f) electrochemical impedance spectroscopy of Co3O4 and Co3O4@α-Fe2O3 NAs anodes. The inset shows equivalent electrical circuit model for the impedance analysis. | ||
The cyclic voltammetry (CV) measurement was then carried out to study the reactive process. Li metal was used as the counter and reference electrodes. Fig. 7c shows the first three CV curves of the Co3O4@α-Fe2O3 NAs at room temperature in the range of 0.005–3.0 V at a slow scan rate of 0.5 mV s−1. As it can be seen, the cathodic peak (first cycle) located around ∼0.52 V with a shoulder at about ∼0.88 V can be attributed to the formation of a solid electrolyte interface (SEI) layer on the electrode surface, which disappears from the second cycle. The subsequent well-defined anodic peaks are observed at 1.68 V and 2.32 V, indicating the extraction of Li+ in the electrode materials. These results nearly coincide with the voltage plateaus in the galvanostatic discharge–charge curve (Fig. 7a). In addition, another cathodic at ∼0.83 V appears since the second cycle, attributed to the reduction of Fe3+ to Fe0.45 In the anodic polarization process, a broad peak on 2.35 V is obtained in the first cycle and successive cycles, which is ascribed to the oxidation of Fe and Co.45–47 It is worth noting that the CV peaks change little during the subsequent cycles. This means that the electrochemical reaction becomes highly reversible after the first discharge–charge, which is consistent with its good cycling performance as discussed below.
For a better understanding of the superior electrochemical performance of the Co3O4@α-Fe2O3 NAs compared with Co3O4 NAs for lithium energy storage, the electrochemical impedance spectroscopy (EIS) was performed at room temperature with an amplitude of 5.0 mV over the frequency range from 100 kHz to 0.01 Hz (Fig. 7f). The result shows that the Nyquist plot exhibits two distinct parts including a semicircle in the high frequency region and a sloped line in the low frequency region, further demonstrating the long term electrochemical stability of these electrode materials. Comparing to the Co3O4 NAs anodes, the anodes made of Co3O4@α-Fe2O3 NAs exhibit larger slopes and shorter lines in the low frequency region, suggesting faster Li+ diffusion rates and smaller variation of diffusion paths.48,49 The EIS data are analyzed by fitting to an equivalent electrical circuit shown in the inset of Fig. 7f, similar to the circuit employed for other oxide electrodes. The intercepts of the depressed semicircles at the real part axis in the high frequency region indicate the total electrical impedance of charge transfer, illustrated by an equivalent electrical circuit consisting of the resistances of electrolyte, electrode, and the interfacial charge transfer process, where Rs is the electrolyte resistance, Rct is the charge transfer resistance, Cdl is the double layer capacitance, W is the Warburg diffusion resistance, CMO is the pseudo-capacitor of metal oxides, RMO is the internal resistance of metal oxides.49 The Co3O4 and Co3O4@α-Fe2O3 NAs show the charge transfer resistances (Rct) of 22.56 and 13.28 Ω. It is noteworthy that the low Rct values for both the Co3O4@α-Fe2O3 NAs and the Co3O4 NAs are attributed to their primary Co3O4 NAs with good conductivity towards Ni foam substrates and efficient ion transport behavior.28
4. Conclusions
In summary, a novel hierarchical Co3O4@α-Fe2O3 NAs heterostructure was designed by a stepwise, seed-assisted hydrothermal approach. First, the single-crystal, original Co3O4 NAs were grown on Ni foam substrates, and then by subsequent coated a layer of α-Fe2O3 nanosheets on the surface of the Co3O4 nanoneedles. The 1D primary Co3O4 trunks facilitate the ion transfer towards the Ni foam current collector, and α-Fe2O3 nanosheets can provide larger contact area with the electrolyte, thus increasing the lithium ion storage capacity to increase unit capacity. Comparing to pristine Co3O4 NAs and α-Fe2O3 nanosheets, the Co3O4@α-Fe2O3 NAs show good capacity retention with 1045 mA h g−1 after the 100th discharge–charge cycle as anode material for lithium battery, as well as high rate capability. The dramatic improvement in the LIBs performance can be ascribed to the advantages endowed by the well-ordered active nanostructure arrays grown directly on metal substrate materials, such as good contact of the active materials and adhesion with the current collector, large interfacial area for lithium insertion/extraction, and reduced ion diffusion pathways. The synthetic strategy of such nanoarchitecture electrode can be extended to other active oxides, thus creating new opportunities for designing a wide range of high-performance LIBs electrode materials.Acknowledgements
This work is supported by the Singapore National Research Foundation under NRF RF Award no. NRF-RF2010-07, the National Natural Science Foundation of China (no. 60171009 and no. U1204501).Notes and references
- C. Zhou, Y. W. Zhang, Y. Y. Li and J. P. Liu, Nano Lett., 2013, 13, 2078–2085 CrossRef CAS PubMed.
- Y. S. Luo, J. S. Luo, J. Jiang, W. W. Zhou, H. P. Yang, X. Y. Qi, H. Zhang, H. J. Fan, D. Y. W. Yu, C. M. Lid and T. Yu, Energy Environ. Sci., 2012, 5, 6559–6566 CAS.
- W. Q. Zeng, F. P. Zheng, R. Z. Li, Y. Zhan, Y. Y. Li and J. P. Liu, Nanoscale, 2012, 4, 2760–2765 RSC.
- J. Chmiola, C. Largeot, P. L. Taberna, P. Simon and Y. Gogotsi, Science, 2010, 328, 480–483 CrossRef CAS PubMed.
- J. Liu, G. Z. Cao, Z. G. Yang, D. H. Wang, D. Dubois, X. D. Zhou, G. L. Graff, L. R. Pederson and J. G. Zhang, ChemSusChem, 2008, 1, 676–697 CrossRef CAS PubMed.
- Y. Qi, N. Du, H. Zhang, X. Fan, Y. Yang and D. Yang, Nanoscale, 2012, 4, 991–996 RSC.
- M. D. Fleischauer, J. Li and M. J. Brett, J. Electrochem. Soc., 2009, 156, 33–36 CrossRef PubMed.
- J. Jiang, Y. Y. Li, J. P. Liu and X. T. Huang, Nanoscale, 2011, 3, 45–48 RSC.
- L. Li and N. Koshizaki, J. Mater. Chem., 2010, 20, 2972–2978 RSC.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- X. Wang, X. L. Wu, Y. G. Guo, Y. Zhong, X. Cao, Y. Ma and J. Yao, Adv. Funct. Mater., 2010, 20, 1680–1686 CrossRef CAS.
- X. Xia, J. Tu, Y. Zhang, X. Wang, C. Gu, X. Zhao and H. J. Fan, ACS Nano, 2012, 6, 5531–5538 CrossRef CAS PubMed.
- H. Wu, M. Xu, H. Wu, J. Xu, Y. Wang, Z. Peng and G. Zheng, J. Mater. Chem., 2012, 22, 19821–19825 RSC.
- J. Jiang, Y. Y. Li, J. P. Liu, X. T. Huang, C. Z. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- Y. Wang, H. Xia, L. Lu and J. Lin, ACS Nano, 2010, 4, 1425–1432 CrossRef CAS PubMed.
- Y. Qi, H. Zhang, N. Du, C. X. Zhai and D. Yang, RSC Adv., 2012, 2, 9511–9516 RSC.
- X. Wang, X. L. Wu, Y. G. Guo, Y. Zhong, X. Cao, Y. Ma and J. Yao, Adv. Funct. Mater., 2010, 20, 1680–1686 CrossRef CAS.
- K. M. Shaju, F. Jiao, A. Débart and P. G. Bruce, Phys. Chem. Chem. Phys., 2007, 9, 1837–1842 RSC.
- Y. Li, B. Tan and Y. Wu, Nano Lett., 2008, 8, 265–270 CrossRef CAS PubMed.
- X. W. Lou, D. Deng, J. Y. Lee, J. Feng and L. A. Archer, Adv. Mater., 2008, 20, 258–262 CrossRef CAS.
- C. Zhang, J. Chen, Y. Zeng, X. Rui, J. Zhu, W. Zhang, C. Xu, T. M. Lim, H. H. Hng and Q. Yan, Nanoscale, 2012, 4, 3718–3724 RSC.
- J. Jiang, J. P. Liu, X. T. Huang, Y. Y. Li, R. M. Ding, X. X. Ji, Y. Y. Hu, Q. B. Chi and Z. H. Zhu, Cryst. Growth Des., 2010, 10, 70–75 CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- N. Du, H. Zhang, B. D. Chen, J. B. Wu, X. Y. Ma, Z. H. Liu, Y. Q. Zhang, D. R. Yang, X. H. Huang and J. P. Tu, Adv. Mater., 2007, 19, 4505–4509 CrossRef CAS.
- Y. Wang, H. Xia, L. Lu and J. Lin, ACS Nano, 2010, 4, 1425–1432 CrossRef CAS PubMed.
- H. Wu, M. Xu, Y. C. Wang and G. F. Zheng, Nano Res., 2013, 6, 167–173 CrossRef CAS.
- X. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- W. W. Zhou, C. W. Cheng, J. P. Liu, Y. Y. Tay, J. Jiang, X. T. Jia, J. X. Zhang, H. Gong, H. H. Hng, T. Yu and H. J. Fan, Adv. Funct. Mater., 2011, 21, 2439–2445 CrossRef CAS.
- Y. L. Wang, J. J. Xu, H. Wu, M. Xu, Z. Peng and G. F. Zheng, J. Mater. Chem., 2012, 22, 21923–21927 RSC.
- Y. Zhao, J. X. Li, Y. H. Ding and L. H. Guan, Chem. Commun., 2011, 47, 7416–7418 RSC.
- Y. Wang, H. Xia, L. Lu and J. Lin, ACS Nano, 2010, 4, 1425–1432 CrossRef CAS PubMed.
- B. Q. Cao and W. P. Cai, J. Phys. Chem. C, 2008, 112, 680–685 CAS.
- Y. C. Ling, G. M. Wang, J. Reddy, C. C. Wang, J. Z. Zhang and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 4074–4079 CrossRef CAS PubMed.
- T. J. Chuang, C. R. Brundle and D. W. Rice, Surf. Sci., 1976, 59, 413–429 CrossRef CAS.
- M. A. Langell, M. D. Anderson, G. A. Carson, L. Peng and S. Smith, Phys. Rev. B: Condens. Matter, 1999, 59, 4791–4798 CrossRef CAS.
- G. X. Wang, L. Yang, Y. Chen, J. Z. Wang, S. Bewlay and H. K. Liu, Electrochim. Acta, 2006, 51, 4634–4638 CrossRef PubMed.
- G. Bhargava, I. Gouzman, C. M. Chun, T. A. Ramanarayanan and S. L. Bernasek, Appl. Surf. Sci., 2007, 253, 4322–4329 CrossRef CAS PubMed.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS PubMed.
- X. H. Xia, J. P. Tu, Y. Q. Zhang, Y. J. Mai, X. L. Wang, C. D. Gu and X. B. Zhao, RSC Adv., 2012, 2, 1835–1841 RSC.
- X. F. Wang, J. B. Xu, X. J. Yu and K. Xue, Appl. Phys. Lett., 2007, 91(031908), 1–3 Search PubMed.
- C. F. Windisch Jr, G. J. Exarhos and R. R. Owings, J. Appl. Phys., 2004, 95, 5435–5442 CrossRef PubMed.
- D. L. A. de Faria, S. V. Silva and M. T. de Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef CAS.
- X. J. Zhu, Y. Z. Zhu, S. Murali, M. D. Stoller and R. S. Ruoff, ACS Nano, 2011, 5, 3333–3338 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, J. Power Sources, 2001, 97, 235–239 CrossRef.
- H. Wu, M. Xu, Y. C. Wang and G. F. Zheng, Nano Res., 2013, 6, 167–173 CrossRef CAS.
- J. Chen, L. Xu, W. Li and X. L. Gou, Adv. Mater., 2005, 17, 582–586 CrossRef CAS.
- Z. S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- J. Chen, X. H. Xia, J. P. Tu, Q. Q. Xiong, Y. X. Yu, X. L. Wang and C. D. Gu, J. Mater. Chem., 2012, 22, 15056–15061 RSC.
- H. Wu, M. Xu, Y. C. Wang and G. F. Zheng, Nano Res., 2013, 6, 167–173 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47189f |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |