
Prediction of acidity constants of some important selenium oxoacids in aqueous solution by computational techniques†
Abstract
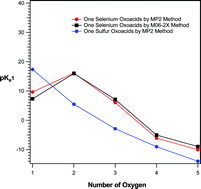
The acidity constants (pKa) of two important series of selenium oxoacids including H2SeOn (n = 1–5) and H2Se2On (n = 1, 3, 4, 6 and 7) were predicted by using two different computational methods for the first time. The calculations were performed using MP2 level of theory employing a very large basis set, 6-311++G(3df,3pd), and a new density functional method, M06-2X, with the same basis set, separately. A new continuum solvation model, SMD, based on the quantum mechanical charge density of a solute molecule interacting with a continuum description of the solvent, was used to account the solvent effects. The calculated values of pKa were corrected using the different correlation equations, reported in the literature, to improve the accuracy of results. Also, the calculated results in this work were compared with the corresponding results related to sulfur oxoacids obtained in our previous work. The same trend was observed for the variation of the acidity constants of selenium oxoacids and corresponding sulfur oxoacids (with the number of oxygen atoms). Comparison of the pKa1 of selenium and sulfur oxoacids showed that the acidity strength of sulfur is higher than selenium while for pKa2, selenium oxoacids are stronger acids than sulfur oxoacids. The predicted values of pKa of selenium oxoacids are important and useful in the different chemical aspects of these compounds in chemistry and biochemistry.
 Please wait while we load your content...
Please wait while we load your content...

