DOI:
10.1039/C3RA47136E
(Paper)
RSC Adv., 2014,
4, 14691-14699
Diverse silver(I) sulfobenzoate coordination polymers and their recycling property as homogeneous catalyst in oxygenation of sulfide†
Received
29th November 2013
, Accepted 6th February 2014
First published on 7th February 2014
Abstract
Four diverse silver(I) coordination polymers, namely {[Ag3(bpmb)2.5(2-sb)(2-Hsb)]·(0.5H2O)}n (1), [Ag2(bpmb)3(2-Hsb)2]n (2), {[Ag2(bpmb)2(3-sb)]·4H2O}n (3), and {[Ag(bpmb)(4-Hsb)]·2H2O}n (4) where bpmb is 1,4-bis(pyrazolyl-methyl)-benzene and sb is sulfobenzoate dianion, have been synthesized and fully characterized by elemental analysis, powder X-ray analysis, IR spectra, TG analysis, fluorescence study, and single crystal X-ray analysis. In these complexes, the bpmb ligand plays the role of bidentate bridge linking with silver ions, while the sb ligands display versatile coordination modes. The combination of the two ligands manages the silver ions to give rise to a variety of coordination networks in 1–4. The accessorial secondary interactions such as hydrogen bonding in complexes 1–4 and aromatic stacking in complexes 1–3 are also helpful for the extension and stabilization of the final supramolecular aggregates. Moreover, the catalytic activity to selective oxidation of methyl phenyl sulfide with H2O2 to sulfoxide also has been explored. Complexes 2 and 4 afforded 98% and 96% conversion with 93% and 90% selectivity for the methyl phenyl sulfoxide in 3 h at 323 K, respectively. In particular, complex 2 is a homogeneous catalyst during the catalytic reaction while it can be recovered by filtration upon cooling and then reused at least four times without losing activity, which is very rare in the oxygenation of sulfides.
Introduction
Crystal engineering of construction design and the rational synthesis of coordination polymers based on the interactions of metal ions with organic ligands,1 is a popular area of current research2 in recent decades owing to their intriguing aesthetic structures and topological features as well as their potential applications in magnetism, electric conductivity, molecular adsorption, heterogeneous catalysis, nonlinear optics and fluorescent materials.3–7 Coordination polymers developed from silver ions and heterocyclic nitrogen ligands continue to attract attention due to the coordinative sites of the d10 silver(I) ion varying from 2 to 6 and its various geometries (linear, trigonal, tetrahedral, trigonal, pyramidal, and octahedral).8–11 The supramolecular chemistry of Ag(I) coordination polymers represents a dynamic and thriving field which abounds with various supramolecular forces such as metal–ligand, metal–π, and metal–metal interactions, hydrogen bonds, π–π stacking, and anion interactions.10,12–14 Therefore the crystallization of Ag(I) complexes would depend on the delicate balance of thermodynamic and kinetic contributions concerning synergetic supramolecular interactions, which may account for the fact that the structures and topologies of Ag(I) complexes can be astonishingly varied even if they have similar ligands.12,15,16 Sulfobenzoates with sulfonate and carboxylate groups are useful synthetic synthons which can develop multimodal and polydentate coordinative systems. The silver coordination complexes with several sulfobenzoates have also been reported in our group and in some of these cases interesting polymeric systems have been structurally characterized.16,17 However, reports on the coordination complexes based on the sulfobenzoates with various N-donor ligands are still limited.18,19 In addition, our team has recently focused on the catalytic activity of the metal–organic coordination complexes with sulfobenzoates as ligands and explored the catalytic activity of the oxidation reaction of methyl phenyl sulfide (MPS).20 Sulfoxides are important in organic chemistry,21–23 owing to their versatile usage as intermediates or products in the synthesis of pharmaceuticals, agrochemicals, and other fine chemicals.24,25 Some silver salts including AgNO3 have been reported to convert sulfides into sulfoxides.26 However, these salts suffer from low activity even when maintined for a long time at a higher temperature. Our initial research on transition metal sulfobenzoate complexes exhibited their catalytic activity on the oxidation of MPS20 and showed the sulfobenzoate ligand can promote the solubility of the complexes. Therefore the combination of sulfobenzoate and silver is expected to have catalytic activity on MPS. Here, this paper describes synthesis, crystal structures, and catalytic properties of four complexes, namely {[Ag3(bpmb)2.5(2-sb)(2-Hsb)]·(0.5H2O)}n (1), [Ag2(bpmb)3(2-Hsb)2]n (2), {[Ag2(bpmb)2(3-sb)]·4H2O}n (3), and {[Ag(bpmb)(4-Hsb)]·2H2O}n (4) (sb = sulfobenzoate dianion, bpmb = 1,4-bis(pyrazolyl-methyl)-benzene).
Results and discussion
Reaction chemistry and XRD study for complexes 1–4
To get these complexes, different methods and starting materials have been explored. For complexes 1–3, CH3COOAg was used as the starting material through a combination of a hydrothermal method and solvent evaporation, while complex 4 was directly synthesized from the cooling solution of the reaction system using AgNO3 as the starting material. Another point we should emphasize is the different molar ratios in the reaction. If the molar ratio of CH3COOAg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2-H2sb:bpmb was fixed at 2:2:1at 393 K, complex 1 was obtained, while when the ratio of starting materials was changed to 1:1:1 with the other conditions kept the same, complex 2 could be achieved. The synthesis of complexes 1 and 2 is only different in the molar ratio and the quantity of starting materials. The environmental synthetic condition for complex 1 was at a lower concentration for starting materials and lower molar ratio of Ag:bpmb. Therefore, these two factors led to the preparation of two diverse complexes. Complex 3 was obtained by a procedure similar to that of 2 except using 3-NaHsb instead of 2-H2sb. Compared with 1–3, complex 4 was synthesized under a higher temperature of 423 K. Moreover complexes 1–3 were obtained in the solvent mixture of water and acetonitrile, while complex 4 was obtained only using water as the solvent in the synthetic process.
:2-H2sb:bpmb was fixed at 2:2:1at 393 K, complex 1 was obtained, while when the ratio of starting materials was changed to 1:1:1 with the other conditions kept the same, complex 2 could be achieved. The synthesis of complexes 1 and 2 is only different in the molar ratio and the quantity of starting materials. The environmental synthetic condition for complex 1 was at a lower concentration for starting materials and lower molar ratio of Ag:bpmb. Therefore, these two factors led to the preparation of two diverse complexes. Complex 3 was obtained by a procedure similar to that of 2 except using 3-NaHsb instead of 2-H2sb. Compared with 1–3, complex 4 was synthesized under a higher temperature of 423 K. Moreover complexes 1–3 were obtained in the solvent mixture of water and acetonitrile, while complex 4 was obtained only using water as the solvent in the synthetic process.
Based on the above synthetic information, we can conclude that the starting materials, pressure, temperature, solvent, and synthetic conditions, especially molar ratio are so important in the synthesis of complexes 1–4, which may be mainly attributed to the coordination property of the sb ligands, since sb has two different functional groups and their coordination abilities are largely influenced by the external stimuli such as synthetic conditions.
These complexes were separated only as single crystals without any powder. The purities of these complexes were further confirmed by XRD analysis, in which the experimental XRD patterns are consistent with those obtained from the single crystal data at room temperature (See ESI,† Fig. S1–S8). The only difference in reflection intensities between the simulated and experimental patterns was due to the variation in the preferred orientation of the powder samples during the collection of the experimental XRD data.
X-ray crystallography
Complex 1 is a two-dimensional structure constructed from Ag(I), bpmb, 2-Hsb−, 2-sb2−, and lattice water molecules. There are three silver ions in an asymmetric unit as shown in Fig. 1 and two 2-sb ligands exist in two forms, one is fully deprotonated and the other is partly deprotonated (a and b in Scheme 1). The coordination spheres of all Ag ions can be described in a tetrahedral geometry. Ag1 adopts two nitrogen atoms from two bpmb ligands and two oxygen atoms from the carboxyl group (O1) of the 2-Hsb− and the sulfonate group (O10) of the 2-sb2−. Ag2 is coordinated to one nitrogen atom from one bpmb ligand and three oxygen atoms from the carboxyl group (O1 and O2) of the 2-Hsb− and the sulfonate group (O8) of 2-sb2− ligands. Ag3 is a tetrahedron coordination geometry completed by two nitrogen atoms from two bpmb and two oxygen atoms from the μ3-carboxyl group and the μ2-sulfonate group (O3) of the 2-sb2− ligand. Ag1 and Ag2 are bridged by the sulfonate group of the 2-Hsb− and the carboxyl group of the 2-sb2− with the Ag⋯Ag distance of 3.6833(6) Å. The Ag2 and Ag3 are bridged by the carboxyl of the 2-sb2− with the Ag⋯Ag distance of 4.0428(5) Å. The 2-sb2− ligand coordinates to Ag1, Ag2 and Ag3 through the carboxyl and sulfonate groups. The bpmb ligands link both Ag2 and its symmetric one and both Ag1 and Ag3. Therefore 2-sb2− and bpmb extend the structure into a 2-D architecture (Fig. S9†).
 |
| | Fig. 1 ORTEP view of the asymmetric unit of complex 1 with numbering scheme and probabilities drawn at 30%. The H atoms and lattice water molecule are omitted for clarity. Symmetry codes, (i): −1 + x, 1 + y, z; (ii): 1 + x, y, z. | |
 |
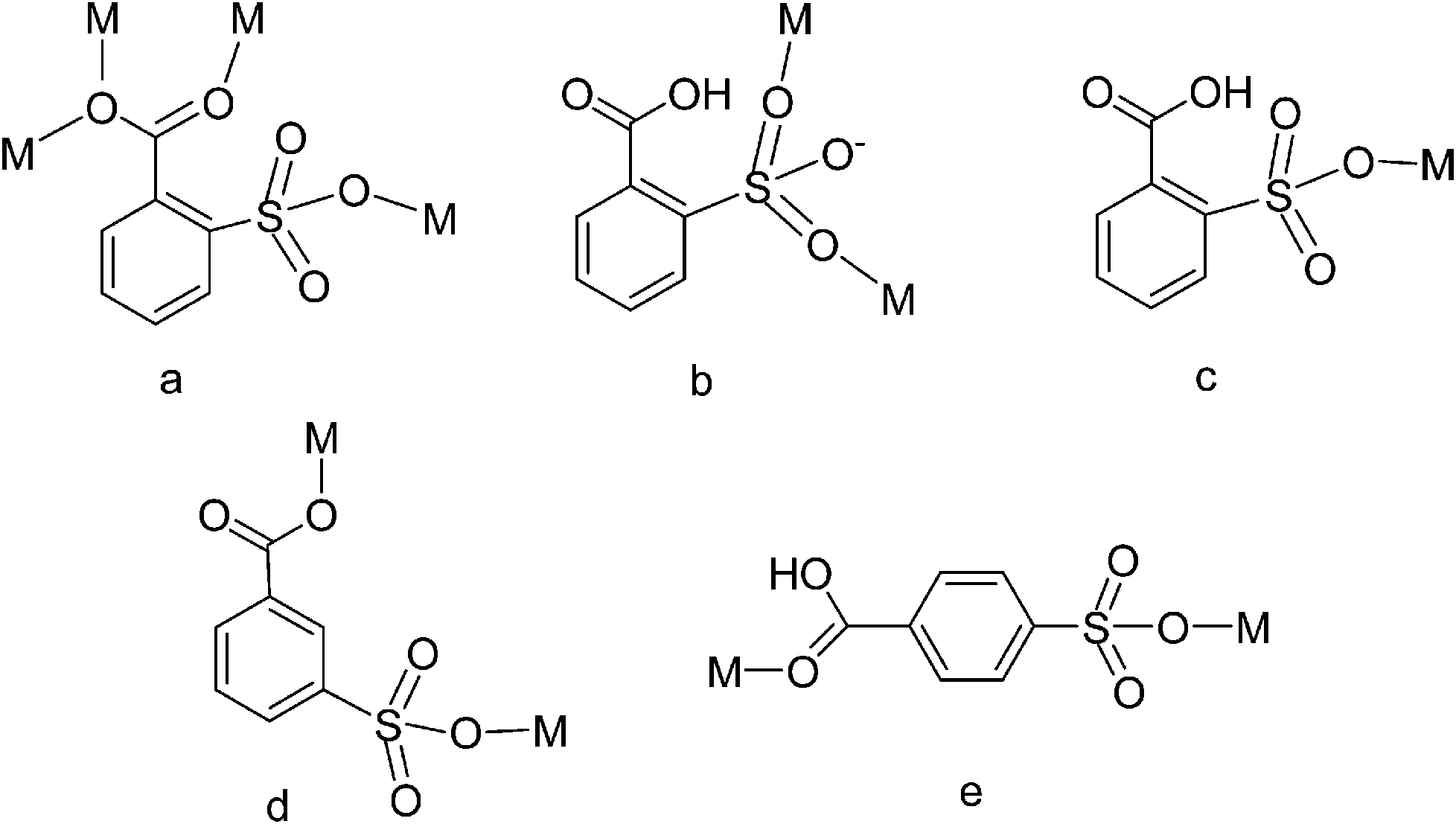
| | Scheme 1 The coordination modes of sb ligands in complexes 1–4. | |
The single crystal X-ray analysis revealed that complex 2 consists of Ag(I), bpmb, and 2-Hsb− (Fig. 2 and c in Scheme 1), which is very different from that of 1. In the asymmetric unit, there is only one Ag(I) ion and its coordination geometry is a tetrahedron completed by three N donors from three bpmb ligands and one O atom from the sulfonate group (O3) with a longer distance of 2.8195(19) Å (Fig. 2 and Table 1). In complex 2, the bpmb ligands adopt a bridging bidentate coordination fashion, forming a 2-D framework (Fig. S10†).
 |
| | Fig. 2 ORTEP view of the asymmetric unit of complex 2 with numbering scheme. The thermal ellipsoids are drawn at 30% probability. The weak bond is represented as an open line. The H atoms are omitted for clarity. | |
Table 1 Selected bond lengths (Å) and anglesa (°)
| Complex 1 |
| Symmetry codes: in 1: (i): –1 + x, 1 + y, z; (ii): 1 + x, y, z; in 3, (i), −x, 2 − y, 1 − z; (ii): x, 1 − y, 1 − z; (iii): −x, 1 − y, −z; in 4, x, −1 + y, z. |
| Ag1–O1 |
2.349 (3) |
Ag1–O10 |
2.705 (3) |
| Ag1–N1 |
2.215 (3) |
Ag1–N10ii |
2.190 (3) |
| Ag2–O1 |
2.904 (5) |
Ag2–O2 |
2.183 (3) |
| Ag2–O8 |
2.588 (4) |
Ag2–N5 |
2.178 (3) |
| Ag3–O2 |
2.396 (3) |
Ag3–O3 |
2.542 (3) |
| Ag3–N4i |
2.275 (4) |
Ag3–N7 |
2.209 (3) |
| O1–Ag1–O10 |
101.26 (11) |
O1–Ag1–N1 |
96.03 (15) |
| O1–Ag1–N10ii |
112.23 (15) |
O10–Ag1–N1 |
96.14 (11) |
| O10–Ag1–N10ii |
100.30 (11) |
N1–Ag1–N10ii |
143.45 (12) |
| O1–Ag2–O2 |
48.09 (11) |
O1–Ag2–O8 |
91.50 (12) |
| O1–Ag2–N5 |
142.27 (11) |
O2–Ag2–O8 |
111.73 (13) |
| O2–Ag2–N5 |
156.81 (14) |
O8–Ag2–N5 |
90.38 (12) |
| O2–Ag3–O3 |
86.37 (12) |
O2–Ag3–N4i |
93.86 (12) |
| O2–Ag3–N7 |
131.44 (12) |
O3–Ag3–N4i |
120.95 (12) |
| O3–Ag3–N7 |
85.52 (11) |
N4i–Ag3–N7 |
130.61 (12) |
| |
| Complex 2 |
| Ag1–O3 |
2.8195 (19) |
Ag1–N1 |
2.300 (2) |
| Ag1–N3 |
2.245 (2) |
Ag1–N5 |
2.269 (2) |
| O3–Ag1–N1 |
98.55 (7) |
O3–Ag1–N3 |
101.11 (7) |
| O3–Ag1–N5 |
82.38 (7) |
N1–Ag1–N3 |
115.81 (8) |
| N1–Ag1–N5 |
112.15 (8) |
N3–Ag1–N5 |
130.64 (8) |
| |
| Complex 3 |
| Ag1–O1 |
2.299 (3) |
Ag1–O1w |
3.001 (4) |
| Ag1–N1 |
2.257 (3) |
Ag1–N3 |
2.234 (3) |
| Ag2–O4 |
2.612 (4) |
Ag2–N5 |
2.113 (3) |
| Ag2–N8ii |
2.109 (3) |
|
|
| O1–Ag1–O1w |
94.04 (11) |
O1–Ag1–N1 |
113.04 (11) |
| O1–Ag1–N3 |
120.92 (12) |
O1w-Ag1-N1 |
107.38 (11) |
| O1w-Ag1-N3 |
76.67 (11) |
N1–Ag1–N3 |
125.53 (11) |
| O4–Ag2–N5 |
100.40 (13) |
O4–Ag2–N8ii |
89.64 (12) |
| N5–Ag2–N8ii |
169.24 (13) |
|
|
| |
| Complex 4 |
| Ag1–O2i |
2.883 (3) |
Ag1–O3 |
2.612 (3) |
| Ag1–N1 |
2.151 (3) |
Ag1–N3 |
2.148 (3) |
| O2i-Ag1-O3 |
87.26 (12) |
O2i-Ag1-N1 |
86.20 (10) |
| O2i-Ag1-N3 |
100.22 (11) |
O3–Ag1–N1 |
112.03 (12) |
| O3–Ag1–N3 |
86.61 (12) |
N1–Ag1–N3 |
160.69 (12) |
Complex 3 consists of silver ions, 3-sb2−, bpmb, and lattice water molecules. There are two silver ions in an asymmetric unit shown in Fig. 3. Ag1 is surrounded by two nitrogen atoms from two bpmb ligands and two oxygen atoms from one carboxyl group and one water molecule with a longer distance of 3.001(4) Å (Table 1). Ag2 has a T-shaped coordination geometry completed by two nitrogen atoms from two bpmb ligands and one oxygen atom from one sulfonate group (O4). The 3-sb2− ligand bridges Ag1 and Ag2 through carboxyl and sulfonate groups (d in Scheme 1). The bpmb ligands also act as a bridging ligand, connecting to two silver ions. The molecular structure of 3 is a 2-D architecture (Fig. S11†).
 |
| | Fig. 3 ORTEP view of the asymmetric unit of complex 3 with the numbering scheme. The thermal probability is drawn at 30%. H atoms and lattice water molecules are omitted for clarity. The weak bond is represented as open line. Symmetry codes: (i), −x, 2 − y, 1 − z; (ii): x, 1 − y, 1 − z; (iii): −x, 1 − y, −z. | |
The asymmetry unit of complex 4 contains one silver ion, one 4-Hsb− ligand, one bpmb ligand, and two lattice water molecules (Fig. 4 and e in Scheme 1). Ag1 is in a distorted tetrahedral geometry completed by two nitrogen atoms from two bpmb ligands and two oxygen atoms from two 4-Hsb− ligands [one is from carboxyl group and the other is from sulfonate group (O3)] with a longer distance of 2.883(3) Å (Table 1). Both bpmb and 4-Hsb− ligands act as bridging linkers to extend the structure into a 2-D layer (Fig. S12†).
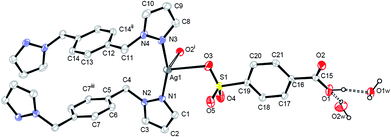 |
| | Fig. 4 ORTEP view of the asymmetric unit of complex 4 with numbering scheme. The probabilities are drawn at 30%. The weak bond is represented as an open line and the hydrogen bonds are represented as dashed lines. Symmetry codes, (i): x, −1 + y,z; (ii): 2 − x, 1 − y, 1 − z; (iii): 1 − x, 1 − y, 1 − z. | |
Hydrogen bonds, C–H⋯aromatic interactions, and C–H⋯O interactions exist in all these complexes. The aromatic stacking effects only exist in complexes 1–3 and in 2 this interaction is weak.
Thermal stability
Thermo-gravimetric analysis (TGA) was conducted to study the stability of the four complexes (Fig. 5). The TGA curve for complex 1 has three degradation steps in the range 42–750 °C. The first gradual weight loss of 0.70% occurs between 42 and 175 °C (calculated 0.68%), corresponding to the loss of a half water molecule per formula unit. In the temperature range 186–312 °C complex 1 lost weight of 29.58% without a clear platform, corresponding to the release of the two coordinated sb ligands (calculated 30.18%). Compared with 1, complex 2 is stable up to ca. 210 °C, and the first-step weight loss of 14.72% in the temperature range 210–270 °C for the release of one sb ligand (calculated 15.11%) without a clear platform, and then a successive decomposition step occurs, which is attributed to loss of the sb and bpmb ligands. For complex 3, the first weight loss of 7.47% occurs between 25 and 120 °C (calculated 7.47%), corresponding to the loss of the four guest water molecules. Then, complex 3 started to decompose at 190 °C. Complex 4 began to lose weight of 6.22% (calculated 6.17%) in the temperature range 25–150 °C, which is accounted for by the liberation of two lattice water molecules. And complex 4 decomposed at 240 °C. The thermal stability analysis for the four complexes shows some differences. Complexes 1, 3, and 4 contain solvents and they lost weight at low temperatures. Complex 2 has no solvent and the decomposition temperature lies at 210 °C. Complexes 1 and 2 contain the same ligands of bpmb and 2-sulfobenzoate, while the existence of the forms of 2-sulfobenzoate ligands in both complexes are different and their coordination modes are also different. Therefore complex 2 and desolvented 1 have somewhat different decomposition temperatures. Desolvented complexes 1, 3 and 4 have different sulfobenzoate ligands and these sulfobenzoate ligands exhibit different coordination modes, leading to some different decomposition temperatures.
 |
| | Fig. 5 TGA curves of complexes 1–4. | |
UV spectra analysis
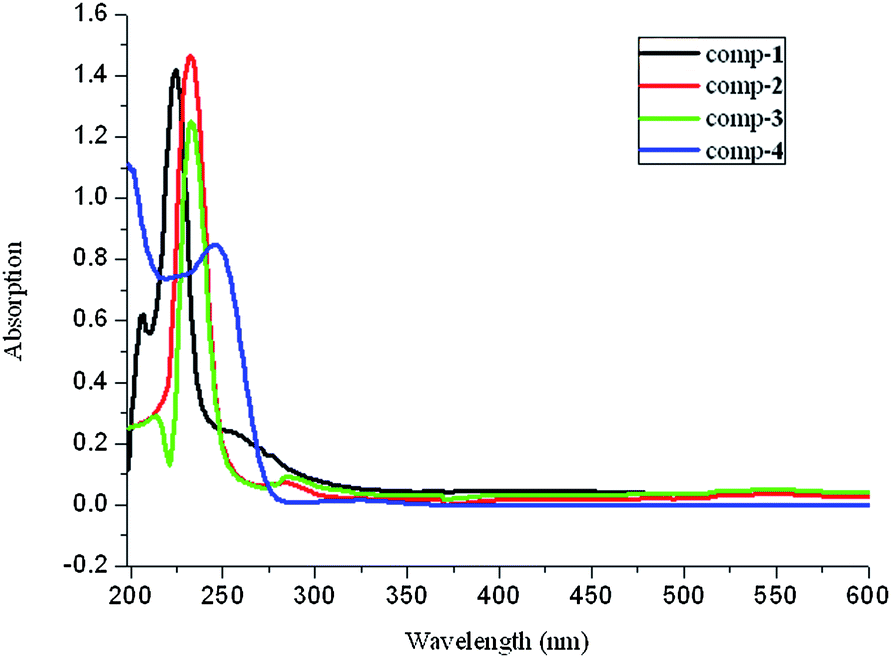
The UV-vis spectra were measured in methanol with a concentration of 2.22 × 10−5 mol L−1. These complexes have more intensive absorptions than those of free ligands, indicating that the coordination of the ligands can enhance the absorption (Fig. 6 and Table 2). The absorptions of complexes 1–2 with the 2-sb ligand are slightly stronger than those of 3–4, while the absorption of the 2-H2sb ligand is weaker than that of the 3-NaHsb and 4-KHsb ligands, indicating that the coordination has a significant influence on the absorptions of 2-H2sb. The shifts of adsorption peaks in four complexes are controlled by many factors, such as solvent effects, conjugation, super-conjugation, steric hindrance, and the ligand type in complexes. The peaks near 220 nm for complexes 1–3 are red-shifted compared with those of the corresponding ligands. However, complex 4 has a distinct blue shift in comparison with 4-KHsb at 234 nm. This may be ascribed to the fact that the para-position of sulfonate in complex 4 plays an important role in better conjugation although the electronic effect of ortho-position of 1 and para-position of 4 are similar. Better conjugation results in a lower energy level of the basic state of electrons. Consequently the energy level difference is bigger and a blue shift takes place in complex 4. The bands at 253 nm in 1, 283 nm in 2, and 285 nm in 3 that are the results of metal-to-ligand charge transfer (MLCT) are observed.27
 |
| | Fig. 6 UV-vis spectra of complexes 1–4 in methanol. | |
Table 2 UV-vis absorption spectral data for complexes 1–4 in methanol with a concentration of 2.22 × 10−5 mol L−1
| Complex |
λmax/nm (ε/dm3 mol−1 cm−1) |
| 1 |
253 (1.09 × 104) |
224 (6.38 × 104) |
207 (2.79 × 104) |
| 2 |
283 (0.33 × 104) |
232 (6.59 × 104) |
|
| 3 |
285 (0.42 × 104) |
233 (5.63 × 104) |
|
| 4 |
246 (3.82 × 104) |
222 (3.820 × 104) |
201 (4.92 × 104) |
Fluorescence properties
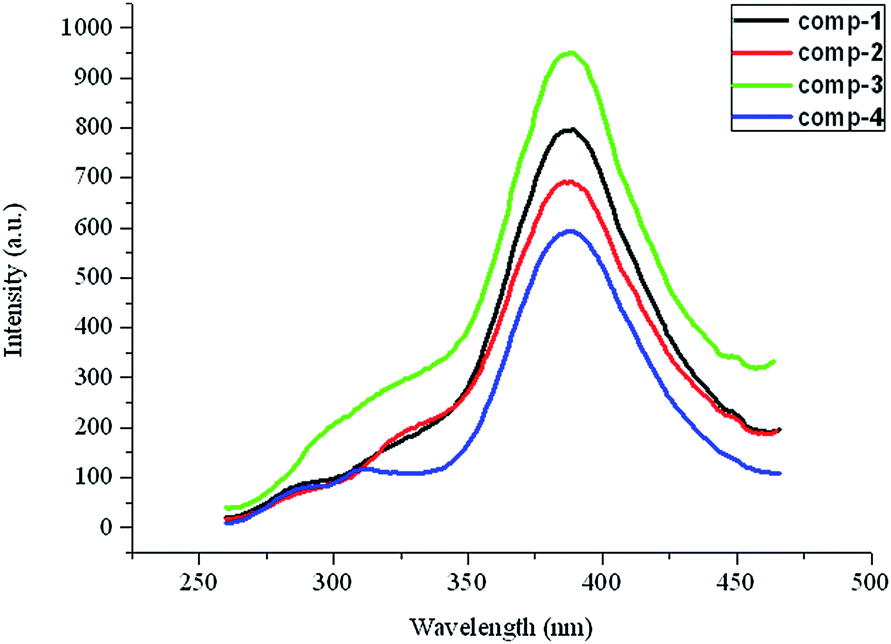
The solid-state fluorescent properties of complexes 1–4 have been investigated at room temperature, as depicted in Fig. 7. The maximum emissions for these complexes occur at 389 nm (λex = 240 nm), which is similar to those of the acidic and neutral ligands, showing that the coordination does not affect the fluorescent emissive position dramatically. The fluorescent strengths of the four complexes are all obviously lower than the corresponding ligands, and especially they almost quench the emission at 289 nm, at which the bpmb ligand has a strong emission. The reason for the intensity reduction or even quenching probably is due to the introduction of the metal ions and solvent molecules, which play an important role in weakening the fluorescent emission.28–30 Meanwhile, the weak interactions, especially the hydrogen-bonding interactions, also play an important role in weakening the fluorescence intensity of the supermolecules.31,32 It is also worth noting that the different coordination modes of 2-sb may result in small different intensities in complexes 1 and 2.
 |
| | Fig. 7 Solid-state emission spectra for complexes 1–4. | |
Catalytic activities
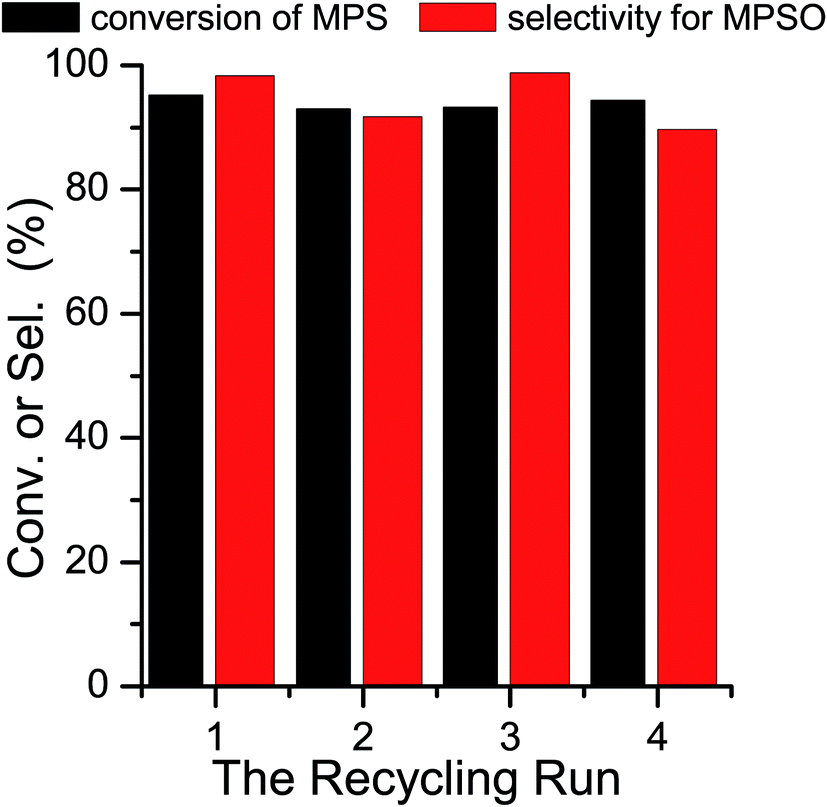
The catalytic activities of four complexes in the oxidation of methyl phenyl sulfide (MPS) at 50 °C were investigated (Scheme 2). The results indicated that the structures of the complexes are largely related to the catalytic activity of the oxidation of MPS. Catalysts 2 and 4 are more active than those of 1 and 3 (Fig. 8). The acid can promote the conversion of the oxidation of MPS.33 Therefore, the 2-H2sb (2 mol% vs. MPS) was introduced into the reaction system, and the experimental results are shown in Fig. 9. Complexes 2 and 4 both are highly efficient and selective catalysts (Table 3). Moreover, complex 2 can be reused at least four times (Fig. 10) without losing activity and shows no significant change in XRD patterns compared with the fresh one (Fig. S13–14†). These four complexes are composed of silver, bpmb, and sulfobenzoate ligands, and they exhibited large different catalytic activity on the oxidation of MPS. Complex 2 has the highest catalytic activity, but it should be used as a catalyst with the 2-H2sb. Their catalytic activity largely depends on the several factors, such as the amount of oxidant, solvent, temperature, and the nature of the catalyst. Complexes 1 and 3 are nearly insoluble in ethanol solvent, while complexes 2 and 4 are both gradually dissolved in the mixture, with the reaction going proceeding as a homogenous catalysis at the reaction temperature (50 °C), and after cooling to the room temperature complex 2 could be separated out by simple filtration (the recycling quantity is about 80%). Therefore complexes 1 and 3 can be used as heterogeneous catalysts, while complexes 2 and 4 can be used as homogeneous catalysts. Complex 4 could not be precipitated from the reaction solution. As a result, only complex 2 can be reused. The 2-H2sb as a catalyst performed below 80% conversion and could not be promoted further. The combination of the neat complex 2 and 2-H2sb can exhibit a cooperative effect with a conversion of 98% due to the 2-H2sb providing the acidic environment. The electron-rich sulfur atom of the MPS undergoes electrophilic oxidation by H2O2 to produce the sulfoxide and sulfone, which suggests that the acidic environment is beneficial for the oxidation of MPS, and the titration experiment further confirmed the formation of intermediates with proton.34 In the early stages, people used strong acid as a reagent to oxidize MPS, but such a system brought about environmental pollution. Therefore people now use H2O2 as an oxidant for the oxidation of MPS under suitable catalysts. These four complexes could not provide some acidity and the combination of complex 2 and 2-H2sb with moderate acidity can significantly exert the catalytic activity. In our previous reports on Ru and Pd sulfobenzoate complexes,35 the 2-sb complexes also exhibited higher catalytic activity than those of 3-sb or 4-sb metal complexes, suggesting that the ortho position of the sulfo group provides a better conjugation leading to its dominance in catalyzed oxidation. The complex 2 as a catalyst under the cooperative effect by 2-H2sb in the oxidation of MPS is the first recyclable homogeneous catalyst with excellent catalytic activity.
 |
| | Scheme 2 The oxidation of MPS. | |
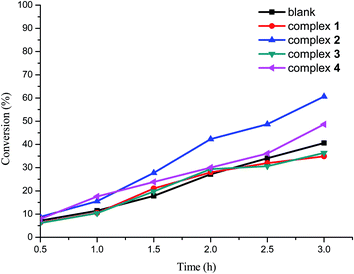 |
| | Fig. 8 The conversion of MPS vs reaction time plot for the oxidation of MPS by complexes 1–4. Blank experimental conditions: 0.5 mmol of MPS and 1.5 mmol of H2O2 were mixed in 5 mL of ethanol at 50 °C. The representative GC chromatograms are presented in ESI (Fig. S15–21).† | |
 |
| | Fig. 9 The conversion of MPS vs reaction time plot for the oxidation of MPS by complexes 1–4 in the presence of 2-H2sb. | |
Table 3 The results of the oxidation of methyl phenyl sulfide by complexes 1–4
| Complex |
Time/h |
Conversion/% |
Selectivity for MPSO/% |
| 1 |
3 |
35 |
86 |
| 2 |
3 |
77 |
94 |
| 3 |
3 |
37 |
91 |
| 4 |
3 |
53 |
90 |
| 1 + 2-H2sb |
3 |
57 |
93 |
| 2 + 2-H2sb |
3 |
98 |
93 |
| 3 + 2-H2sb |
3 |
43 |
90 |
| 4 + 2-H2sb |
3 |
96 |
91 |
| 2-H2sb |
3 |
79 |
96 |
| Blank |
3 |
40 |
82 |
 |
| | Fig. 10 Catalytic activities of four recycling runs for complex 2. | |
Conclusion
In summary, we have successfully synthesized four silver sulfobenzoate coordination polymers with bpmb ligand. The variable positions of sulfonate groups on benzene rings gave rise to different coordination polymers and variable chemical properties. The 2-sb ligand yields more abundant coordination modes. Complexes 2 and 4 demonstrated high efficiency and good selectivity in the oxidation of MPS to the corresponding sulfoxide using H2O2 as the oxidant. The results indicate that these catalysts have higher catalytic activities than those silver salts or other transition metal catalysts. The sulfonate group can promote solubility in polar solvents, and this specific character largely helps the complex can act as homogenous catalyst, but it can be recovered by simple cooling of complex 2, which is an excellent example of recycling homogeneous catalysts.
Experimental section
General information
Chemicals were purchased from commercial sources and were used without further purification. C, H, and N elemental analyses were carried out on a Perkin-Elmer analyzer model 1110. The IR spectra were obtained on a Nicolet Nexus 470 infrared spectrophotometer in KBr pellets in the 400–4000 cm−1 region. The fluorescence spectra were measured in a powder sample using a SHIMADZU RF-540 spectrometer at room temperature. Thermogravimetric analyses (TGA) were carried out using a Delta Series TA-SDT Q600 under nitrogen flow over the temperature range room temperature to 800 °C at a heating rate of 10°C min−1 with Al2O3 crucibles. The UV-vis spectra were obtained on a Varian Bio 2550 UV-visible spectrophotometer in CH3OH at room temperature. 1H and 13C NMR spectra were recorded on a Bruker NMR AVANCEIII 400 or 500 spectrometer. The powder X-ray diffractions were measured by Rigaku D/MaX 2550 PC with Cu-Kα radiation. The GC data were recorded on a Fuli Gas Chromatography equipped with a DB-5 capillary column. All the standard substances used in the GC were purchased from Alfa Aesar. The retention times for the assignment of GC peaks for sulfide, sulfoxide and sulfone are about 4.6 min, 7.8 min, and 8.5 min, respectively under following conditions. Detector: FID, 220 °C; sample injector: splitless, 200 °C; column temperature: temperature programming with heating rate of 15 °C min−1, initial temperature of 100 °C for 3 min and final temperature of 200 °C for 2 min.
Synthesis of 1,4-bis (pyrazolyl-methyl)-benzene (bpmb)
1,4-Bis (pyrazolyl-methyl)-benzene (bpmb) was synthesized by one step according to the literature reference.36 Pyrazole (9.032 g, 0.13 mol), α,α′-dichloro-p-xylene (10.52 g, 0.065 mol), and tetrabutyl ammonium bromide (1.024 g, 3 mmol) were mixed in benzene (600 mL), and then 100 mL 40% NaOH solution was added under stirring. The mixture was refluxed for about 24 h. After removing benzene using a rotator, yellow powder was obtained, which was dissolved in ethyl acetate and re-crystallized at room temperature. After further re-crystallization twice, a white crystalline product was obtained. 1H NMR [DMSO-d6, 25 °C, 500 MHz; δ(ppm)]: 7.79 (d, J = 5.0 Hz, 2H), 7.44 (d, J = 5.0 Hz, 2H), 7.16 (s, 4H), 6.25 (t, J = 5.0 Hz, 2H), 5.29 (s, 4H).
Synthesis of {[Ag3(bpmb)2.5(2-sb)(2-Hsb)]·(0.5H2O)}n (1)
A mixture of CH3COOAg (0.032 g, 0.20 mmol), 2-H2sb (0.042 g, 0.20 mmol), bpmb (0.029 g, 0.12 mmol), distilled water (13 mL) and acetonitrile (3 mL) was placed in a 30 mL Teflon lined stainless-steel reactor and heated at 393 K for two days. After cooling to the room temperature, a clear solution was obtained and then filtered. The resulting solution was set in the dark for evaporation. Two days later, colorless block-shaped crystals were collected by filtration. Yield: 67%. Anal. calcd (%) for C98H90N20Ag6S4O21: C, 44.28; H, 3.38; N, 10.54. Found: C, 44.32; H, 3.33; N, 10.49. IR (KBr pellet, cm−1): 3519 (m), 3106 (s), 2937(m), 1731(s), 1587 (s), 1560 (s), 1517(m), 1438 (s), 1424(s),1405 (s), 1370 (s), 1271 (s), 1258(s), 1183 (s), 1095 (s), 1080(s), 1060 (s), 1014(s), 979 (m), 919(w), 843 (m), 808(m), 786 (s), 756(s), 727 (s), 614 (s), 567(m). 1H NMR [DMSO-d6, 25 °C, 400 MHz; δ(ppm)]: 14.17 (s, 1H), 7.92–7.79 (m, 7H), 7.47–7.39 (m, 11H), 7.18 (s, 10H), 6.31 (t, J = 5.0 Hz, 5H), 5.33 ppm (s, 10H). 13C NMR [DMSO-d6, 25 °C, 400 MHz; δ(ppm)]: 140.0, 136.7, 131.0, 129.3, 127.8, 126.8, 105.7, 54.4.
Synthesis of [Ag2(bpmb)3(2-Hsb)2]n (2)
The synthesis of 2 was similar to that described for 1, except for the different molar ratios of the starting materials and heating time. A mixture of CH3COOAg (0.065 g, 0.40 mmol), 2-H2sb (0.084 g, 0.40 mmol), bpmb (0.095 g, 0.40 mmol), distilled water (13 mL) and acetonitrile (3 mL) was placed in a 30 mL Teflon lined stainless-steel reactor and heated at 393 K for 36 h. After cooling to room temperature, a clear solution was obtained and then filtered. The resulting solution was allowed to evaporate in the dark for two days. Then colorless plate-like crystals were collected by filtration. Yield: 76%. Anal. calcd (%) for C56H52N12Ag2S2O10: C, 50.53; H, 3.76; N, 12.63. Found: C, 50.30; H, 3.83; N, 12.52. IR (KBr pellet, cm−1): 3519 (m), 3117 (s), 2935 (m), 2656 (w), 1716 (s), 1589 (w), 1518 (s), 1438 (s), 1424 (s), 1400 (s), 1358 (m), 1286 (s), 1258 (s), 1238 (s), 1187 (s), 1165 (s), 1141 (s), 1093 (s), 1078 (m), 1060 (m), 1021 (m), 1002 (m), 976 (m), 890 (w), 849 (w), 767 (s), 745 (s), 728 (m), 617 (s), 570 (m). 1H NMR [DMSO-d6, 25 °C, 400 MHz; δ(ppm)]: 14.17 (s, 2H), 7.86–7.74 (m, 10H), 7.57–7.47 (m, 10H), 7.16 (s, 12H), 6.30 (t, J = 2.0 Hz, 6H), 5.31 (s, 12H). 13C NMR [DMSO-d6, 25 °C, 400 MHz; δ(ppm)]: 167.6, 144.2, 139.5, 136.9, 131.1, 130.8, 130.6, 129.7, 127.7, 126.6, 105.7, 54.4.
Synthesis of {[Ag2(bpmb)2(3-sb)]·4H2O}n (3)
This complex was prepared similarly to 2 except for the use of 3-NaHsb instead of 2-H2sb. After cooling to room temperature, a clear solution was obtained and then filtered. The resulting solution was allowed to evaporate in the dark. Two days later, colorless block crystals were collected by filtration. Yield: 75%. Anal. calcd (%) for C35H40N8Ag2SO9: C, 43.58; H, 4.15; N, 11.62. Found: C, 43.87; H, 4.01; N, 11.55. IR (KBr pellet, cm−1): 3432 (s), 3116 (m), 3090 (m), 2934 (w), 1600 (s), 1560 (s), 1516 (s), 1437 (s), 1424 (s), 1399 (s), 1385 (s), 1358 (s), 1284 (s), 1229 (s), 1185 (s), 1168 (s), 1093 (s), 1050 (s), 1037 (s), 1020 (m), 975 (m), 915 (m), 808 (m), 767 (s), 745 (s), 668 (m), 616 (s), 485 (m). 1H NMR [DMSO-d6, 25 °C, 400 MHz; δ(ppm)]: 8.31 (m, J = 1.6 Hz, 1H), 7.92–7.88 (m, 5H), 7.68–7.66 (m, 1H), 7.51–7.50 (m, 4H), 7.33–7.30 (m, 1H), 7.16 (s, 8H), 6.33 (t, J = 2.0 Hz, 4H), 5.34 (s, 8H). 13C NMR [DMSO-d6, 25 °C, 400 MHz; δ(ppm)]: 170.1, 147.6, 140.1, 136.7, 136.5, 131.1, 129.6, 127.8, 127.1, 127.0, 127.0, 106.0, 54.4.
Synthesis of {[Ag(bpmb)(4-Hsb)]·2H2O}n (4)
A mixture of AgNO3 (0.032 g, 0.20 mmol), 4-KHsb (0.047 g, 0.20 mmol), bpmb (0.045 g, 0.20 mmol) and distilled water (15 mL) was placed in a 30 mL Teflon lined stainless-steel reactor and heated at 423 K for two days. After cooling to room temperature, colorless needle-shaped crystals were collected by filtration. Yield: 64%. Anal. calcd (%) for C21H23N4AgSO7: C, 43.27; H, 3.94; N, 9.61. Found: C, 43.03; H, 3.86; N, 9.51. IR (KBr pellet, cm−1): 3478 (s), 3424 (s), 3134 (m), 3116 (m), 3090 (m), 1701(s), 1654 (m), 1636 (m), 1513 (m), 1437 (m), 1424 (m), 1400 (s), 1358 (m), 1285 (s), 1185 (s), 1136 (m), 1094 (m), 1033 (m), 1011 (s), 975 (m), 915 (w), 767 (s), 718 (m), 636 (s), 485 (m). 1H NMR [DMSO-d6, 25 °C, 400 MHz; δ(ppm)]: 12.98 (s, 1H), 7.91–7.86 (m, 4H), 7.71–7.69 (m, 2H), 7.48–7.47 (m, 2H), 7.16 (s, 4H), 6.30 (t, J = 2.0 Hz, 2H), 5.32 (s, 4H). 13C NMR [DMSO-d6, 25 °C, 400 MHz; δ(ppm)]: 167.0, 152.2, 139.5, 136.9, 130.6, 128.9, 127.7, 125.7, 105.7, 54.4.
X-ray crystallographic determination
Crystallographic data were collected at 295 K on an Oxford Diffraction Xcalibur CCD diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The frames were integrated with the CrysAlisPro package and the data were corrected for absorption using the program CrysAlisPro.37 The structures were solved by direct methods using the program SHELXL-97.38 All non-hydrogen atoms were refined with anisotropic thermal parameters by full-matrix least-squares calculations on F2 using the program SHELXL-97. The graphics were drawn by the ORTEP and Mercury.39,40 Details of the crystal data and structure refinements for the four complexes are listed in Table 4.
Table 4 Crystallographic data and refinement parameters for the complexes 1–4
| Complex |
1 |
2 |
3 |
4 |
| Empirical formula |
C98H90N20O21Ag6S4 |
C28H26N6O5AgS |
C35H40N8O9Ag2S |
C21H23N4O7AgS |
| Mr |
2659.41 |
666.49 |
964.56 |
583.37 |
| Crystal color/shape |
Colorless/block |
Colorless/plate |
Colorless/block |
Colorless/needle |
| Crystal system, space group |
Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Triclinic, P |
Triclinic, P |
Triclinic, P |
| Crystal size |
0.22 × 0.26 × 0.29 |
0.15 × 0.18 × 0.42 |
0.23 × 0.26 × 0.29 |
0.25 × 0.13 × 0.08 |
| a/Å |
10.6768 (4) |
10.4087 (4) |
10.6331 (3) |
8.2280 (4) |
| b/Å |
12.6832 (4) |
11.5583 (5) |
11.3717 (3) |
10.4465 (5) |
| c/Å |
19.6770 (9) |
12.1619 (6) |
18.0884 (5) |
13.3444 (7) |
| α/° |
84.345 (3) |
88.778 (4) |
98.097 (1) |
92.427 (1) |
| β/° |
75.994 (3) |
72.109 (4) |
98.286 (1) |
90.631 (1) |
| γ/° |
76.864 (3) |
80.521 (4) |
113.971 (1) |
94.105 (1) |
| Volume (Å3), Z |
2515.04 (17), 1 |
1372.73 (11), 2 |
1928.99 (9), 2 |
1142.92 (10), 2 |
| Calculated density (g·cm−3) |
1.756 |
1.612 |
1.661 |
1.695 |
| Absorption coefficient (mm−1) |
1.310 |
0.861 |
1.133 |
1.024 |
| F (000) |
1332 |
678 |
976 |
592 |
| Measured reflections |
15742 |
8719 |
19941 |
9446 |
| Unique reflections |
8940 |
4869 |
6820 |
4005 |
| Observed reflections |
6935 |
4263 |
6009 |
3413 |
| θ rang/° |
3.2-25.1 |
3.0-25.1 |
1.2-25.1 |
1.5-25.1 |
| Parameters |
695 |
370 |
520 |
322 |
| Goodness-of-fit on F2 |
1.030 |
1.000 |
1.065 |
1.090 |
| R1 and ωR2 (I > 2σ(I)) |
0.040, 0.090 |
0.032,0.076 |
0.038, 0.107 |
0.032, 0.095 |
| R1 and ωR2 (all data) |
0.058, 0.099 |
0.040, 0.082 |
0.044, 0.117 |
0.042, 0.118 |
| Largest peak and hole (e Å−3) |
0.948, −0.668 |
0.855, −0.503 |
1.173, −0.748 |
0.527, −0.636 |
Catalytic oxidation of methyl phenyl sulfide
The synthesized complex as a catalyst (0.015 mmol) was mixed with 5 mL ethanol in a 25 mL round-bottom glass-reactor, and then the methyl phenyl sulfide (0.5 mmol) was added. The mixture was kept at 323 K under stirring, and the hydrogen peroxide (1.5 mmol) was then introduced as the oxidant. At the same time the initial reaction time was recorded. The experiments were performed by two methods. One is in the absence of the 2-H2sb while the other is in the presence of 2-H2sb (0.0021 g, 0.01 mmol). The reaction progress was monitored by GC. Assignments of products were made by comparison with authentic samples. All the reactions were run at least in duplication.
Acknowledgements
The authors thank the National Natural Science Foundation of China (grant no. 21073157).
Notes and references
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed; M. Du, C. P. Li, C. S. Liu and S. M. Fang, Coord. Chem. Rev., 2013, 257, 1282–1305 CrossRef; R. Patra, H. M. Titi and I. Goldberg, Cryst. Growth Des., 2013, 13, 1342–1349 Search PubMed.
- S. H. Park, S. Y. Lee, K. M. Park and S. S. Lee, Acc. Chem. Res., 2012, 45, 391–403 CrossRef CAS PubMed; Y. Y. Xu, Y. Y. Xing, X. Y. Duan, Y. Z. Li, H. Z. Zhu and G. J. Meng, CrystEngComm, 2010, 12, 567–572 RSC.
- X. L. Tong, H. L. Lin, J. H. Xin, F. Liu, M. Li and X. P. Zhu, J. Nanomater., 2013, 2013, 616501 Search PubMed; M. Clemente-Leon, E. Coronado, C. Marti-Gastaldo and F. M. Romero, Chem. Soc. Rev., 2011, 40, 473–497 RSC; D. Braga and F. Grepioni, Acc. Chem. Res., 2000, 33, 601–608 CrossRef CAS PubMed.
- N. C. Burtch, H. Jasuja, D. Dubbeldam and K. S. Walton, J. Am. Chem. Soc., 2013, 135, 7172–7180 CrossRef CAS PubMed; K. Sumida, D. Stuck, L. Mino, J. D. Chai, E. D. Bloch, O. Zavorotynska, L. J. Murray, M. Dinca, S. Chavan, S. Bordiga, M. Head-Gordon and J. R. Long, J. Am. Chem. Soc., 2013, 135, 1083–1091 CrossRef PubMed; J. R. Li, Y. G. Ma, M. C. McCarthy, J. Sculley, J. M. Yu, H. K. Jeong, P. B. Balbuena and H. C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef; A. C. McKinlay, R. E. Morris, P. Horcajada, G. Ferey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef PubMed.
- S. Nakagaki, G. K. B. Ferreira, G. M. Ucoski and K. A. D. D. Castro, Molecules, 2013, 16, 7279–7308 CrossRef CAS PubMed; C. D. Wu and W. B. Lin, Angew. Chem., Int. Ed., 2007, 46, 1075–1078 CrossRef PubMed; D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48(41), 7502–7513 CrossRef PubMed.
- A. O. Ibrahim, Y. F. Zhou, F. L. Jiang, L. Chen, X. J. Li, W. T. Xu, O. O. E. Onawumi, O. A. Odunota and M. C. Hong, Eur. J. Inorg. Chem., 2011,(32), 5000–5005 CrossRef CAS; I. Goldberg, CrystEngComm, 2008, 10, 637–645 RSC; J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1480 RSC.
- H. L. Jiang and Q. Xu, Chem. Commun., 2011, 47, 3351–3370 RSC; B. Chen, L. Wang, Y. Xiao, F. R. Fronczek, M. Xue, Y. Cui and G. Qian, Angew. Chem., Int. Ed., 2009, 48, 500–503 CrossRef CAS PubMed.
- A. A. Massoud, V. Langer, Y. M. Gohar, M. A. M. Abu-Youssef, J. Janis, G. Lindberg, K. Hansson and L. Ohrstrom, Inorg. Chem., 2013, 52, 4046–4060 CrossRef CAS PubMed; J. Malberg, T. Wiegand, H. Eckert, M. Bodensteiner and R. Wolf, Chem. - Eur. J., 2013, 19, 2356–2369 CrossRef PubMed; D. Venkataraman, Y. Du, S. R. Wilson, K. A. Hirsch, P. Zhang and J. S. Moore, J. Chem. Educ., 1997, 74, 915–918 CrossRef.
- M. Maekawa, T. Miyazaki, K. Sugimoto, T. Okubo, T. Kuroda-Sowa, M. Munakata and S. Kitagawa, Dalton Trans., 2013, 42, 4258–4266 RSC; L. Tei, V. Lippolis, A. J. Blake, P. A. Cooke and M. Schroder, Chem. Commun., 1998, 23, 2633–2634 RSC.
- F. R. Knight, R. A. M. Randall, L. Wakefield, A. M. Z. Slawin and J. D. Woollins, Dalton Trans., 2013, 42, 143–154 RSC; A. N. Khlobystov, A. J. Blake, N. R. Champness, D. A. Lemenovskii, A. G. Majouga, N. V. Zyk and M. Schroder, Coord. Chem. Rev., 2001, 222, 155–192 CrossRef CAS.
- H. R. Khavasi and B. M. M. Sadegh, Cryst. Growth Des., 2012, 12, 4798–4804 Search PubMed; X. P. Li, J. Y. Zhang, M. Pan, S. R. Zheng, Y. Liu and C. Y. Su, Inorg. Chem., 2007, 46, 4617–4625 CrossRef CAS PubMed.
- C. L. Chen, B. S. Kang and C. Y. Su, Aust. J. Chem., 2006, 59, 3–18 CrossRef CAS; C. A. Hollis, S. R. Batten and C. J. Sumby, Cryst. Growth Des., 2013, 13, 2350–2361 Search PubMed; X. F. Zheng and L. G. Zhu, J. Mol. Struct., 2013, 1039, 1–7 CrossRef.
- C. Y. Su, Y. P. Cai, C. L. Chen, M. D. Smith, W. Kaim and H. C. zur Loye, J. Am. Chem. Soc., 2003, 125, 8595–8613 CrossRef CAS PubMed; B. Li, S. Q. Zang, H. Y. Li, Y. J. Wu and T. C. W. Mak, J. Organomet. Chem., 2012, 708, 112–117 CrossRef.
- M. C. Brandys and R. J. Puddephatt, J. Am. Chem. Soc., 2002, 124, 3946–3950 CrossRef CAS PubMed; B. Notash, N. Safari and H. R. Khavasi, CrystEngComm, 2012, 14, 6788–6796 RSC; J. L. Gulbransen and C. M. Fitchett, CrystEngComm, 2012, 14, 5394–5397 RSC.
- X. F. Zheng and L. G. Zhu, Cryst. Growth Des., 2009, 9, 4407–4417 CAS.
- X. F. Zheng and L. G. Zhu, Polyhedron, 2011, 30, 666–675 CrossRef CAS.
- X. H. Miao and L. G. Zhu, Dalton Trans., 2010, 39, 1457–1459 RSC.
- P. Thuery, Inorg. Chem., 2013, 52, 435–447 CrossRef CAS PubMed; B. F. Hoskins, R. Robson and D. A. Slizys, J. Am. Chem. Soc., 1997, 119, 2952–2953 CrossRef; S. Song, X. Li and Y. H. Zhang, Dalton Trans., 2013, 42, 10409–10412 RSC.
- P. Thuery, Cryst. Growth Des., 2011, 11, 5702–5711 Search PubMed; P. Thuery, Cryst. Growth Des., 2012, 12, 1632–1640 Search PubMed; S. Chawla, M. Arora, K. Nattinen, K. Rissanen and J. V. Yakkmi, Polyhedron, 2004, 23, 3007–3019 CrossRef CAS; X. F. Li, Y. B. Huang and R. Cao, Dalton Trans., 2012, 41, 6195–6205 RSC.
- X. H. Miao and L. G. Zhu, CrystEngComm, 2009, 11, 2500–2509 RSC.
- N. Khiar, V. Valdivia, A. Salvador, A. Chelouan, A. Alcudia and I. Fernandez, Adv. Synth. Catal., 2013, 355, 1303–1307 CrossRef CAS; N. Salam, P. Mondal, J. Mondal, A. S. Roy, A. Bhaumik and S. M. Islam, RSC Adv., 2012, 2, 6464–6477 RSC.
- E. Wojaczyńska and J. Wojaczyński, Chem. Rev., 2010, 110, 4303–4356 CrossRef CAS PubMed; W. L. Li, Z. G. Xie and X. B. Jing, Catal. Commun., 2011, 16, 94–97 CrossRef.
- J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347, 19–31 CrossRef CAS; A. Toutchkine and E. L. Clennan, J. Org. Chem., 1999, 64, 5620–5625 CrossRef PubMed.
- H. L. Holland, Nat. Prod. Rep., 2001, 18, 171–181 RSC.
- I. Fernandez and N. Khiar, Chem. Rev., 2003, 103, 3651–3705 CrossRef CAS PubMed.
- R. Das and D. Chakraborty, Synthesis, 2011, 2, 277–280 Search PubMed.
- M. K. Nazeeraddin, S. M. Zakeeruddin and K. Kalyanasundaran, J. Phys. Chem., 1993, 97, 9607–9612 CrossRef CAS; X. Y. Qian, J. L. Song and J. G. Mao, Inorg. Chem., 2013, 52, 1843–1853 CrossRef PubMed; A. Spangenberg, J. P. Malval, H. Akdas-Kilig, J. L. Fillaut, F. Stehlin, N. Hobeika, F. Morlet-Savary and O. Soppera, Macromolecules, 2012, 45, 1262–1269 CrossRef.
- C. Seward, J. Chan, D. Song and S. Wang, Inorg. Chem., 2003, 42, 1112–1120 CrossRef CAS PubMed; H. H. Zhang, W. Dou, W. S. Liu, X. L. Tang and W. W. Qin, Eur. J. Inorg. Chem., 2011, 5, 748–753 CrossRef.
- K. Akhbari, A. Morsali and L. G. Zhu, J. Mol. Struct., 2008, 891, 132–137 CrossRef CAS.
- M. C. Aragoni, M. Arca, F. Demartin, F. A. Devillanova, F. Isaia, A. Garau, V. Lippolis, F. Jalali, U. Papke, M. Shamsipur, L. Tei, A. Yari and G. Verani, Inorg. Chem., 2002, 41, 6623–6632 CrossRef CAS PubMed.
- S. L. Zheng, M. Gembicky, M. Messerschmidt, P. M. Dominiak and P. Coppens, Inorg. Chem., 2006, 45, 9281–9289 CrossRef CAS PubMed.
- S. L. Zheng and P. Coppens, Cryst. Growth Des., 2005, 5, 2050–2059 CAS.
- W. T. Hu and L. G. Zhu, Chin. J. Inorg. Chem., 2013, 29, 1109–1114 CAS.
- M. R. Maurya, B. Singh, P. Adao, F. Avecilla and J. C. Pessoa, Eur. J. Inorg. Chem., 2007, 5720–5734 CrossRef CAS.
- W. T. Hu and L. G. Zhu, J. Coord. Chem., 2013, 66, 3045–3057 CrossRef CAS.
- E. A. Nudnova, A. S. Potapov, A. I. Khlebnikov and V. D. Ogorodnikov, Russ. J. Org. Chem., 2007, 43, 1698–1702 CrossRef CAS.
- CrysAlisPro, Version 1.171.33.52, Oxford Diffraction Ltd., 2009 Search PubMed.
- G. M. Sheldrick, SHELXL-97, program for crystal structure refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra of complexes 1–4. Representative GC chromatograms. 1H NMR for bpmb. 1H NMR and 13C NMR for complexes 1–4. Extended hydrogen-bonding patterns for complexes 1–4. XRD patterns and simulated ones by single crystal data of complexes 1–4 and XRD patterns of complex 2 after the recycle of catalytic reaction. CCDC [958100–958103]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra47136e |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.