
Catalytic behavior of supported Ru nanoparticles on the (101) and (001) facets of anatase TiO2†
Abstract
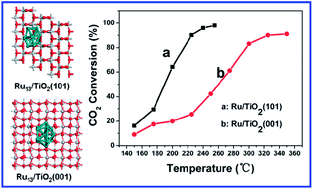
Ru/TiO2 heterogeneous catalysts were prepared by immobilizing Ru nanoparticles onto the (101) and (001) facets of anatase TiO2 substrate, and the influence of metal–support interactions on the catalytic behavior of Ru/TiO2 towards CO2 methanation was studied from the viewpoint of electronic structure. Structural investigations based on temperature-programmed reduction (TPR) and X-ray photoelectron spectroscopy (XPS) indicate that a stronger metal–support interaction occurs between Ru and (101) facet in contrast to the Ru and (001) one. This gives rise to an enhancement in CO2 adsorption as well as spill-over hydrogen at the interface of Ru/TiO2(101), accounting for its largely enhanced catalytic activity towards CO2 methanation. In addition, a theoretical study based on density functional theory (DFT) calculations reveals that the Ru nanoparticles supported on the (101) plane have a relatively lower activation energy for CO dissociation (the rate-determining step), which results in their high activity toward CO2 methanation reaction.
 Please wait while we load your content...
Please wait while we load your content...

