Gold nanoparticles supported on the periodic mesoporous organosilica SBA-15 as an efficient and reusable catalyst for selective oxidation of silanes to silanols†
Lina Maab,
Wenguang Lengb,
Yaopeng Zhaob,
Yanan Gao*b and
Hongdong Duan*a
aSchool of Chemistry and Pharmaceutical Engineering, Qilu University of Technology, Western University Science Park, Daxue Road, Jinan 250353, Shandong, P.R. China. E-mail: hdduan67@gmail.com; Fax: +86-531-89631215; Tel: +86-531-89631215
bDalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: ygao@dicp.ac.cn; Fax: +86-411-84379992; Tel: +86-411-84379992
First published on 6th January 2014
Abstract
Gold nanoparticles are confined and stabilized within the channels of SBA-15 through the poly(ionic liquid) brushes that are anchored onto the pore walls of SBA-15. The supported gold catalyst exhibited remarkably high catalytic activities for selective oxidation of silanes into silanols using water as an oxidant without the use of organic solvents.
Supported gold catalysts have recently attracted great attention for oxidation reactions since the seminal work of Haruta et al.1 The authors showed that nanosized gold nanoparticles (NPs) adsorbed on the metal-oxide exhibited high catalytic activity for the oxidation of CO. Since then, interest in gold supported catalysts has grown exponentially and gold catalysts have been applied to a variety of reactions.2 In particular, the small-sized gold NPs have been considered a crucial criteria for the high catalytic activities.3 However, one big problem when using small NPs is that they are thermodynamically susceptible to agglomeration which leads to the formation of larger inactive particles. A possible method to solve the problem is to develop the solid supports in order to increase the catalyst's stability and to facilitate the catalyst recovery.4
Among the various reactions, the selective oxidation of silanes is a type of important reaction for the production of silanols which have been widely used as building blocks for silicon-containing polymers5 and nucleophilic coupling partners.6 Traditionally, silanols are synthesized using various strong oxidizing agents such as permanganate,7 ozone,8 silver salts,9 peracids,10 dioxiranes,11 or osmium tetroxide12 However, a large amount of siloxanes and toxic by-products are generally formed and the synthesis generates a large amount of environmentally damaging waste.13 An alternative topic in this field is to develop the heterogeneous catalysts that can provide a straightforward and fruitful route to selective oxidation of silanes into silanols using water as an oxidant under mild conditions. However, so far only few elegant examples have appeared in the literatures, including hydroxyapatite-supported silver or gold NPs by Kaneda and co-workers,14,15 nanoporous gold by Asao and co-workers16 and carbon nanotubes-supported gold nanohybrids by John et al.17 Therefore, there still remains a great challenge to develop new reusable catalysts for the selective oxidation of silanes under mild conditions.
Recent studies have shown that the excellent catalytic performance of gold NPs is not only determined by the size and shape of the NPs but also depends on the interaction between gold NPs and support interface.18 To prevent the agglomeration of nanosized gold NPs during reactions, a series of studies on stabilizing NPs by alkyl chains attached through thiol bonds to the surface of the gold were attempted.18 However, the strong interaction between the thiol group and the surface of the gold NPs may alter their catalytic properties profoundly.19 Even a weak interaction such as aromatic group with gold NPs through π-electrons appears to be sufficient to increase the catalyst's stability and to improve its performance.13 To this end, Schrinner et al.20 reported the synthesis of stable Pt NPs embedded in a dense layer of polyelectrolyte chains that are affixed onto the spherical polystyrene cores. The Pt NPs carry no group stabilizing their surface, so they exhibit a high catalytic activity in hydrogenation reactions in the aqueous phase. However, filtering off NPs after use in catalysis still remains a problem. Also, spherical polystyrene cores are susceptible to be dissolved in organic solvents, which will limit the application of the catalyst.
Here, we report that gold NPs confined and stabilized within the novel versatile poly(ionic liquid), poly(1-(4-vinylbenzyl)-3-methylimidazolium chloride) brushes that are anchored onto the pore walls of SBA-15 (namely SBA–PIL–Au composite catalyst) exhibit remarkably high catalytic activities for selective oxidation of silanes into silanols. The advantages of this way to generate gold NPs are obvious: because of the confinement of the channels, the growth of gold NPs is significantly inhibited and at the same time, gold NPs generated within the PIL brushes carry no group stabilizing their surface, both of which could keep the high catalytic activity of original nanoscaled gold particles. Also, the oxidation of silanes only uses water as an oxidant without the use of organic solvents – a substantial environmental advantage. In addition, SBA-15 provides high specific surface area and excellent dispersion for metal NPs. To the best of our knowledge, this is the first catalytic system that confined and stabilized metal NPs by poly(ionic liquid) brushes within the periodic mesoporous silica.
The preparation of SBA–PIL–Au nanohybrid catalyst (see the ESI† for details) started with the surface modification of SBA-15 with the initiator, followed by the atom transfer radical polymerization (ATRP) of ionic liquid monomer. [AuCl4]− ions are immobilized as counterions within the poly(ionic liquid) brushes through the ion exchange. Subsequent reduction with NaBH4 under mild conditions gave SBA–PIL–Au nanohybrids (Fig. 1). FT-IR spectra and TGA measurements confirmed the successful modification of SBA-15 step by step.21 Elemental analysis showed that the gold loading on SBA–PIL–Au was 2.3 wt%, higher than the previous reports.16,22 The XRD patterns, N2 adsorption isotherms,21 and transmission electron microscopy (TEM) in Fig. 2a revealed that the SBA–PIL–Au remained highly ordered 2-dimensional hexagonal mesostructure of maternal SBA-15. Moreover, TEM also showed that the gold NPs were highly dispersed within the modified SBA-15 support with a mean diameter of approximately 2.5 nm (Fig. 2b).
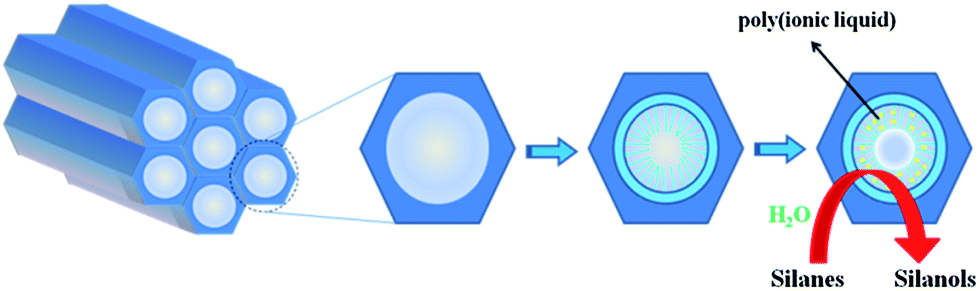 | ||
| Fig. 1 Schematic representation of the SBA–PIL–Au nanohybrids. | ||
 | ||
| Fig. 2 TEM images of (a) SBA–PIL and (b) SBA–PIL–Au. | ||
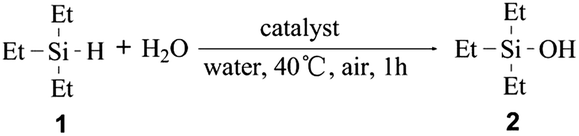
The catalytic performance of the SBA–PIL–Au nanohybrids was tested in the oxidation of silanes with water. We first explore the oxidation of triethylsilane (1) that was selected as a model substrate. The results of the oxidation reaction of 1 over various catalysts were summarized in Table 1. Oxidation of 1 occurred quantitatively to afford triethylsilanol (2) in over 99% conversion without the formation of the disiloxane (Table 1, entry 1). The use of either Au2O3 or HAuCl4 instead of SBA–PIL–Au catalyst resulted in moderate yields of 2, but with the formation of undesirable disiloxane (Table 1, entries 6 and 7). No silanol products were obtained when only using the maternal SBA-15 or under blank conditions (Table 1, entries 8 and 9). To determine whether the oxidation occurs on the supported gold NPs, we stopped the reaction at 57% conversion of 1, and removed the catalyst by centrifugation. Au leaching in the supernatant was not observed by inductively coupled plasma spectrometer (ICP) measurement. Further stirring of the filtrate under the same oxidation conditions did not give any additional product. Upon adding the catalyst again into the reaction mixture, the reaction proceeded well with 100% conversion after half an hour. These results show that SBA–PIL–Au nanohybrids catalyze the oxidation of triethylsilanes. SBA–PIL–Au could be easily retrieved from the reaction mixture by simple centrifugation or filtration, which overcomes the problem of filtering off NPs after use in catalysis. We also found that the recovered SBA–PIL–Au was reusable without significant loss of activity and selectivity, the yield of 2 could be remained at over 99% during 3 recycles of the catalyst (Table 1, entries 3–5). TEM images and XRD patterns for the reused SBA–PIL–Au catalyst were similar to those of the freshly prepared SBA–PIL–Au,22 which indicates that the size of the gold NPs was not significantly changed during oxidation. Therefore, the SBA–PIL–Au catalyst has excellent reusability. To compare its catalytic efficacy with already reported systems, the oxidation of reactant 1 was catalyzed with 0.4 mol% of the SBA–PIL–Au to reach a striking TON of 1500 and TOF of 9000 h−1. To the best of our knowledge, these values are comparable to ever reported catalysts for silane oxidation, and are approximately two orders of magnitude superior to those of NPs loaded systems.14,16,17 (see Table S2† for detailed comparison).
|
|||
|---|---|---|---|
| Entry | Catalyst | Conv.b (%) | Silanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :disiloxane :disiloxane |
| a Reaction conditions: SBA–PIL–Au (0.034 g, Au: 0.4 mol%), triethylsilane (1.0 mmol), H2O (3.0 mL).b Determined by GC using internal standard technique.c At 4 h, 25 °C. | |||
| 1 | SBA–PIL–Au | >99 | >99:1 |
| 2c | SBA–PIL–Au | >99 | >99:1 |
| 3 | Reuse 1 | >99 | >99:1 |
| 4 | Reuse 2 | >99 | >99:1 |
| 5 | Reuse 3 | >99 | >99:1 |
| 6 | HAuCl4 | >99 | 87:13 |
| 7 | Au2O3 | >99 | 89:11 |
| 8 | SBA-15 | <1 | — |
| 9 | None | <1 | — |
It was found that the solvent had a significant influence on the performance of SBA–PIL–Au in this reaction. As shown in Table 2, it took 1 hour to accomplish the quantitative conversion of 1 to 2 in water, while the conversion was no more than 25% in acetone, ethyl acetate (EA) and THF. The catalytic activity of SBA–PIL–Au decreased in the following order: H2O > THF > EA > acetone. Such a difference can be explained by the dispersion of catalyst in these solvents. Our experiment has shown that SBA–PIL–Au nanohybrids are able to highly disperse in water due to the hydrophilic groups of the poly(ionic liquid) brushes, while the dispersion of the catalyst is quite poor in acetone, ethyl acetate and THF, which results in poor contact between reactants and catalyst, and thus a low reaction efficiency.

In the end, a total of 7 different silanes were also tested and the conversion was determined after a given time. As shown in Table 3, SBA–PIL–Au efficiently catalyzed the oxidation of silanes to their corresponding silanols without the formation of any disiloxane products (Table 3). Triisopropylsilane and tributylsilane with sterically bulky aliphatic groups were also oxidized with quantitative conversion (Table 3, entries 3 and 5). Moreover, it is noted that SBA–PIL–Au is also an effective catalyst for phenylsilanes oxidation (Table 3, entries 6 and 7) except for triphenylsilane (Table 3, entry 8).
| entry | Silane | Silanol | Time (h) | Conv.b (%) | Silanol: disiloxane |
|---|---|---|---|---|---|
| a Reaction conditions: silanes (1.0 mmol), H2O (3.0 mL), SBA–PIL–Au (0.304 g, Au: 0.40 mol%), 40 °C, air.b Determined by GC using internal standard technique. | |||||
| 1 |  |
 |
1 | >99 | >99:1 |
| 2 | 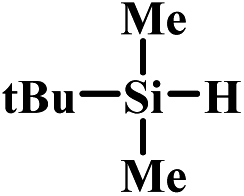 |
 |
5 | >99 | >99:1 |
| 3 | 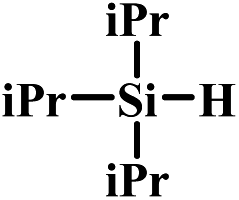 |
 |
24 | >98 | >99:1 |
| 4 |  |
 |
6 | >99 | >99:1 |
| 5 |  |
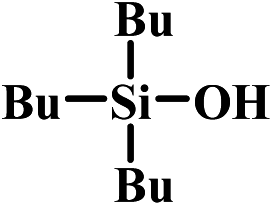 |
6 | >99 | >99:1 |
| 6 |  |
 |
7 | >99 | >99:1 |
| 7 |  |
 |
3 | 99 | >99:1 |
| 8 |  |
 |
24 | 7 | >99:1 |
Based on the above results and reported literatures,23,24 a plausible mechanism is depicted as illustrated in Fig. 3: the first step is the insertion of Si–H bond into Au NPs to generate a silly-nanoparticle conjugate A. This intermediate then reacts with water to produce B, which further breaks into silanol, hydrogen, and regenerated Au NPs through reductive elimination to complete the whole cycle. More studies are still needed to elucidate the exact mechanism.
 | ||
| Fig. 3 Proposed mechanism for the Au NPs catalyzed oxidation of silanes into silanols using water as oxidant. | ||
In summary, gold NPs embedded within the poly(ionic liquid) brushes affixed in a SBA-15 support showed high catalytic activity, excellent selectivity, and reusability for the oxidation of a variety of silanes into their corresponding silanols under mild conditions. Furthermore, it should be noticed that this catalyst also has the following advantages: no requirement for organic solvents, use of water as a clean oxidant, and a wide scope of substrate silanes, including aliphatic silanes. In addition, the catalysts can be designed by attaching functionalized groups on poly(ionic liquid)s toward specific applications. The loading amount of Au is relatively high and can be controlled for a particular purpose. This methodology may be extended to other catalysis systems.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (21273235, 20173189) and the Shandong Provincial Natural Science Foundation, China (ZR2011BM011).Notes and references
- (a) M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405 CrossRef CAS; (b) M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS.
- (a) J. Han, Y. Liu and R. Guo, J. Am. Chem. Soc., 2009, 131, 2060 CrossRef CAS PubMed; (b) A. Corma and P. Serna, Science, 2006, 313, 332 CrossRef CAS PubMed; (c) A. Abad, P. Concepcin, A. Corma and H. García, Angew. Chem., Int. Ed., 2005, 44, 4066 CrossRef CAS PubMed.
- (a) X. Y. Liu, A. Q. Wang, T. Zhang, D. S. Su and C. Y. Mou, Catal. Today, 2011, 160, 103 CrossRef CAS PubMed; (b) A. Q. Wang, Y. P. Hsieh, Y. F. Chen and C. Y. Mou, J. Catal., 2006, 237, 197 CrossRef CAS PubMed.
- (a) C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS PubMed; (b) G. A. Somorjai and J. Y. Park, Angew. Chem., Int. Ed., 2008, 47, 9212 CrossRef CAS PubMed; (c) A. M. Doyle, S. K. Shaikhutdinov and H. J. Freund, Angew. Chem., Int. Ed., 2005, 44, 629 CrossRef CAS PubMed; (d) R. A. VanSanten, Acc. Chem. Res., 2009, 42, 57 CrossRef CAS PubMed.
- V. Chandrasekhar, R. Boomishankar and S. Nagendran, Chem. Rev., 2004, 104, 5847 CrossRef CAS PubMed.
- (a) S. Werner, J. Am. Chem. Soc., 2008, 130, 16382 CrossRef PubMed; (b) S. E. Denmark and D. Wehrli, Org. Lett., 2000, 2, 565 CrossRef CAS PubMed.
- P. D. Lickiss and R. Lucas, J. Organomet. Chem., 1995, 521, 229 CrossRef.
- (a) L. Spialter and J. D. Austin, J. Am. Chem. Soc., 1965, 87, 4406 CrossRef CAS; (b) L. Spialter, L. Pazdernik, S. Bernstein, W. A. Swansiger, G. R. Buell and M. E. Freeburger, J. Am. Chem. Soc., 1971, 93, 5682 CrossRef CAS.
- N. Duffaut, R. Calas and J. C. Macé, Bull. Soc. Chim. Fr., 1959, 1971 CAS.
- L. H. Sommer, L. A. Ull and G. A. Parker, J. Am. Chem. Soc., 1972, 94, 3469 CrossRef CAS.
- (a) W. Adam, R. Mello and R. Curci, Angew. Chem., Int. Ed., 1990, 29, 890 CrossRef; (b) E. G. Rochow and W. F. Gilliam, J. Am. Chem. Soc., 1941, 63, 798 CrossRef CAS; (c) R. O. Sauer, J. Am. Chem. Soc., 1944, 66, 1707 CrossRef CAS; (d) S. M. Sieburth and W. Mu, J. Org. Chem., 1993, 58, 7584 CrossRef CAS.
- D. A. Hrovat, W. T. Borden and J. M. Mayer, Inorg. Chem., 2007, 46, 5212 CrossRef PubMed.
- B. Karimia and F. K. Esfahania, Adv. Synth. Catal., 2012, 354, 1319 CrossRef.
- T. Mitsudome, S. Arita, H. Mori, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2008, 47, 7938 CrossRef CAS PubMed.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2009, 5302 RSC.
- N. Asao, Y. Ishikawa, N. Hatakeyama, Menggenbateer, Y. Yamamoto, M. W. Chen, W. Zhang and A. Inoue, Angew. Chem., Int. Ed., 2010, 49, 10093 CrossRef CAS PubMed.
- J. John, E. Gravel, A. Hagge, H. Y. Li, T. Gacoin and E. Doris, Angew. Chem., Int. Ed., 2011, 50, 7533 CrossRef CAS PubMed.
- (a) T. Mitsudome, S. Arita, H. Mori, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2005, 44, 629 CrossRef PubMed; (b) T. Risse, S. Shaikhutdinov, N. Nilius, M. Sterrer and H. J. Freund, Acc. Chem. Res., 2008, 41, 949 CrossRef CAS PubMed.
- (a) M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Wyman, J. Chem. Soc., Chem. Commun., 1994, 801 RSC; (b) M. J. Hostetler, C. J. Zhong, B. K. H. Yen, J. Anderegg, S. M. Gross, N. D. Evans, M. Porter and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 9396 CrossRef CAS; (c) D. Astruc, F. Lu and R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS PubMed; (d) M. J. Hostetler, J. E. Wingate, C. J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17 CrossRef CAS.
- M. Schrinner, M. Ballauff, Y. Talmon, Y. Kauffmann, J. Thun, M. Möller and J. Breu, Science, 2009, 323, 617 CrossRef CAS PubMed.
- See ESI† for details.
- L. Vradman, M. V. Landau, D. Kantorovich, Y. Koltypin and A. Gedanken, Microporous Mesoporous Mater., 2005, 79, 307 CrossRef CAS PubMed.
- K. Mori, M. Tano, T. Mizugaki, K. Ebitani and K. Kaneda, New J. Chem., 2002, 26, 1536 RSC.
- B. P. S. Chauhana, A. Sarkara, M. Chauhanb and A. Roka, Appl. Organomet. Chem., 2009, 23, 385 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, figures from Fig. S1 to S6, tables from Tables S1 to S2. See DOI: 10.1039/c3ra47047d |
| This journal is © The Royal Society of Chemistry 2014 |