
Probing the folding induction ability of orthanilic acid in peptides: some observations†
Abstract
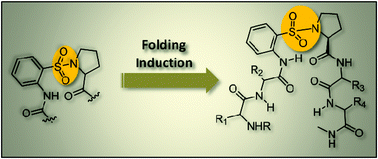
This paper describes the ability of orthanilic acid (2-aminobenzenesulfonic acid, SAnt) to promote folding when introduced in a peptide sequence. Three peptide sequences, containing orthanilic acid (SAnt) with a sulfonamide moiety in the turn segment, have been synthesized in the solution phase using suitable coupling agents, and their structural aspects investigated using NMR and X-ray crystallographic studies. Solid- and solution-state conformational analyses reveal that the peptide sequences containing orthanilic acid in their backbone exist in a folded conformation featuring long-range 15-membered ring H-bonding.
 Please wait while we load your content...
Please wait while we load your content...

