Rapid analysis of drug binding to β-cyclodextrin: part II substituents effect on physicochemical and co-conformational stability of drug/cyclodextrin complex
Ali Aboel Dahab*
Pharmaceutical Sciences Research Division, King's College London, 150 Stamford Street, London SE1 9NH, UK. E-mail: ali.aboel_dahab@kcl.ac.uk; aliadahab@yahoo.co.UK; Fax: +44 (0)2078484462; Tel: +44 (0)2078483944 Tel: +44 (0)7950698740
First published on 20th December 2013
Abstract
In the pharmaceutical industry, the most common application of cyclodextrins (CyDs) is to enhance the solubility, stability, safety and bioavailability of drug molecules. Although, there have been many reports on the complexation of drugs with cyclodextrins, to date, CyDs interaction with drugs is not well understood. The purpose of this work is to show the successful applications of previous work employing a novel, versatile, compact, low-volume, routine apparatus, which consists of a pump, flask and a rotating cell holder for the simultaneous measurements of absorbance and circular dichroism (CD) that allows for the concurrent use of four different pathlengths for binding studies. To show the effect of substituents on drug/CyD binding, benzoic acid as an important precursor for the synthesis of many other organic substances and benzoic acid derivatives were used in this study. Also, an effective novel method for binding titration was employed. The pKa, binding constants, stoichiometry and structural co-conformations of benzoic acid and derivatives/β-CyD complexes were elucidated and determined with accuracy. Substituents type and location show a significant effect on the extent of binding and physicochemical properties of the binding complex. The system proved efficiency, ability to be used routinely and the analysis time was reduced significantly to less than one fourth of the total analysis time used in conventional methods, with possible automation for high-throughput analysis.
Introduction
Molecular recognition is a term used to describe noncovalent interactions between molecules. The field encompasses both theory and experiment of host–guest chemistry and supramolecular chemistry.1 Whether it is sequestration, sensing, crystallization, catalysis, solubility, self-assembly, distribution ratios in immiscible solvents, or drug–receptor interactions, the nature and strength of interactions between molecules creates many of the phenomena of contemporary interest in chemistry, biology, and materials science. Molecular encapsulation of drugs by cyclodextrins (CyDs) is probably the most widely used method to overcome problems associated with drugs, such as poor solubility and unwanted side effects.2,3 The potential guest list for molecular encapsulation in cyclodextrins is quite varied and includes such compounds as straight or branched chain aliphatics, aldehydes, ketones, alcohols, organic acids, fatty acids, aromatics, gases, and polar compounds such as halogens, oxyacids and amines.4 Cyclodextrins also are used as complexing agents to increase drug bioavailability and stability.5,6 For example, cyclodextrins can be used to reduce gastrointestinal drug irritation, convert liquid drugs into microcrystalline or amorphous powder, and prevent drug–drug and drug–excipient interactions.7 Moreover, the encapsulation of drugs by cyclodextrins can be useful since the host molecules may act as concentration-regulating agents, decreasing the adverse side effects at the organ or cellular level and improving the optimal dose of the drug and its frequency of administration.8 Their doughnut-shaped structure, with hydrophilic external faces and hydrophobic inner surface, makes them the most important simple organic compounds capable of forming noncovalently bonded inclusion complexes with a wide variety of drug molecules in aqueous solution. The resulting host–guest systems become more soluble and stable, the stability being a result of the concurrence of different contributions, such as van der Waals interactions, hydrophobic effect, solvent reorganization, and hydrogen bonding.9 This special molecular arrangement of CyDs also accounts for the variety of beneficial effects cyclodextrins have on proteins, which is widely used in pharmacological applications.10 Although inclusion complexes of drug/CyD can assume higher order complexes (e.g. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 drug/CyD), the formation of an inclusion complex often has a stoichiometry of 1:1 interaction, which is illustrated in Fig. 1. Nevertheless, the extent of drug binding to cyclodextrin (binding constant, K) presents a key issue in the whole binding process, it is particularly important in therapeutics, where the pharmacological effect of a drug is directly related to its nature (meaning in free or complexed form).11 The extent of binding varies between drugs and so as the stability of the drug/CyD complex, depending on various factors such as physicochemical and conformational properties of the drugs used. The physicochemical properties of drugs, including their complexation ability, may be greatly affected by the properties of the substituents such as their type, number, and position on the parent drug molecule. In addition, the pH of the medium i.e. the ionisation state of the guest compound has a significant effect on the thermodynamic parameters of the (drug/CyD) complexation reaction. To date, the influence of pH has not been investigated in detail yet, only a few articles are devoted to this problem, using indirect methods such as potentiometry12 and phase-solubility studies.13 However, experimentally determined values of some spectroscopic variables, e.g., fluorescence quantum yield,14,15 absorption coefficient,16,17 chemical shift18,19 and induced circular dichroism (ICD)16 can be used to monitor such reactions.
:1 drug/CyD), the formation of an inclusion complex often has a stoichiometry of 1:1 interaction, which is illustrated in Fig. 1. Nevertheless, the extent of drug binding to cyclodextrin (binding constant, K) presents a key issue in the whole binding process, it is particularly important in therapeutics, where the pharmacological effect of a drug is directly related to its nature (meaning in free or complexed form).11 The extent of binding varies between drugs and so as the stability of the drug/CyD complex, depending on various factors such as physicochemical and conformational properties of the drugs used. The physicochemical properties of drugs, including their complexation ability, may be greatly affected by the properties of the substituents such as their type, number, and position on the parent drug molecule. In addition, the pH of the medium i.e. the ionisation state of the guest compound has a significant effect on the thermodynamic parameters of the (drug/CyD) complexation reaction. To date, the influence of pH has not been investigated in detail yet, only a few articles are devoted to this problem, using indirect methods such as potentiometry12 and phase-solubility studies.13 However, experimentally determined values of some spectroscopic variables, e.g., fluorescence quantum yield,14,15 absorption coefficient,16,17 chemical shift18,19 and induced circular dichroism (ICD)16 can be used to monitor such reactions.
 | ||
| Fig. 1 Binding of a drug with a cyclodextrin to form a 1:1 inclusion complex. | ||
Optical activity measurements, circular dichroism (CD) in particular, offer a convenient way of monitoring absolute stereochemistry and shape in solution at relatively low concentration, mainly with respect to solution variables such as temperature, concentration, pH, and additives. However, when dealing with achiral drugs, which is the case in this study, induced circular dichroism (ICD) can be used for monitoring drug–ligand interactions.20 On the other hand, measurements of absorption alongside CD measurements are important and often necessary. The UV/Vis absorption spectrum of an organic compound is normally composed of only a few broad spectral features, where pathlength/concentration/analysis and wavelength combinations are chosen to optimise measurements. Circular dichroism spectroscopy has often been used to study molecular binding,21 and CD spectra are more generally analysed for molecular structure information, conformation and configuration.22,23 It is desirable to have a complete CD spectrum covering the whole spectral range, however, the selection rules for ordinary absorption and CD are different, e.g. prominent features in a CD spectrum such as n–π* of CO2H may not be apparent in the ordinary absorption. Therefore, to measure a complete CD spectrum while keeping the ordinary absorption in the optimum range (A = 0.5 to 1.5), measurements have to be made in stages with different pathlengths and/or concentrations. Thus, typically the aromatic phenyl chromophore has three major absorption bands with the characteristics: 1Lb (ε275 = 100–1000), 1La (ε210 = 10000), 1E (ε195 = 50000).24 Conventionally, this is likely to be undertaken with three separate scans in three different cells; changes in concentration may have implications for solution studies particularly those involving concentration-dependent equilibria such as binding investigations.25 Therefore, in binding studies, the use of conventional methods is time consuming and prone to errors due to manual withdrawal and addition.
This article describes applications of a novel four-flow cell system that has been recently introduced by us26 for rapid binding analysis using the simultaneous measurement of absorption and circular dichroism.27 The binding of β-CyD as a function of pH to benzoic acid and benzoic acid derivatives as important precursors for the synthesis of many other organic substances (e.g. antimicrobial drugs) were chosen as an example to study the effect of substituents on binding interactions and on the stability of drug/β-CyD complex. The new system was used and the analysis time was reduced to less than one fourth of that for conventional methods. Using an effective approach for binding titration, the extent of binding, stoichiometry and the change of physicochemical properties are estimated with accuracy. The mathematical analysis of the spectroscopic variations in binding titrations and the equations used to estimate the binding constant are explained. Also, the co-conformation of β-CyD and these molecules are assigned and elucidated according to spectroscopic changes upon complexation. The results show the applicability and efficiency of the new system in determining the impact of substituents on physicochemical and co-conformational stability of drug/cyclodextrin complex.
Materials and methods
Materials
NaOH and HCl were purchased from BDH, UK, benzoic acid, and benzoic acid derivatives from Sigma-Aldrich Co. LLC., UK. Water was distilled in-house at the pharmacy department, King's College London.Methods
 | ||
| Fig. 2 Schematic diagram showing the practical principles of the setup for the new system. | ||
The advantages of this system over the conventional methods are that in addition to faster analysis time, it helps avoiding solution loss and concentration errors during the manual transfer of measurement solution from one cell to another particularly small pathlength cells e.g. 0.05 or 0.01 cm cells. Also, it provides efficient method for mixing of solutions with variable speeds that can be controlled by the operating pump. This is predominantly important when a lot of stirring is required as it is the case in binding and pH titration especially when the solutions involved are sensitive to mechanical aggregation and precipitation e.g. protein28 or when dealing with extremely volatile compounds29 which can lead to concentration variations.
The binding of drugs to cyclodextrin as a function of pH potentially requires the consideration of four equilibria:

The pH titrations were performed to determine the pKa values for both the free and the bound drugs to conclude the change of physicochemical properties upon binding with β-cyclodextrin.
For pH values < pH 3.0, 10.0 μl aliquots of 3.0 M HCl need to be added; between pH 3.0 and pH 10.0, 10.0 μl aliquots of 0.1 M NaOH/HCl suffice; above pH 10.0, 3.0 M NaOH is required. Appropriate volumes of drug solutions of appropriate concentrations were added to the titration flask and titrated with the addition of 10 μl aliquots of NaOH/HCl leaving the drug concentration effectively constant. The total volume was ∼2.01–2.1 ml which was sufficient to ensure good mixing by the pump. The solution was mixed thoroughly using the pump and spectra were measured in the normal way at each pH interval with the cell carousel being rotated to change the cell (pathlength).
:15 respectively (solution B), then, pushing the equilibrium to completion with solution A, initially in the titration apparatus titrated with solution B. This is achieved by withdrawing a certain amount of solution A, then adding the same amount from solution B and measuring after each addition (Fig. 3). Alternatively, the titration can start from the fully bound state solution B and progressively reducing the host concentration, however, in this case it is not possible to achieve a completely zero host concentration.
 | ||
| Fig. 3 Schematic diagram showing the most effective ways in binding titration. | ||
As the pKas for these compounds are between 2.0 and 7.0, pH 2.0 was chosen as the lower limit and pH 7.0 as the higher limit to ensure achieving the two state ionisation required for this study.
| host (H) + guest (G) ⇌ [host:guest complex] |
At equilibrium the complex concentration (HG) and free host and guest concentrations (Hf and Gf respectively) are related by:
| Hf + Gf ⇌ HG, HT = Hf + HG and GT = Gf + HG, |
However, based upon Beer's law:
| Aobs = AHf + AGf + AHG | (1) |
| Āobs = εHHf + εGGf + εHGHG | (2) |
| Āobs = εHHT + εGGT + (εHG − εH − εG)HG | (3) |
Combining standard solution equilibria equations for solving for the complex concentration, HG as a function of HT, and substitution of Beer's law equations gives:26
 | (4) |
In eqn (4), Āobs is the dependent variable depending upon three spectroscopic constants (the extinction coefficients of the original species G, H and HG), the association constant (Kass) and the original (total) guest and host concentrations. To obtain a value for the binding constant eqn (4) was entered in the Origin program. A plot of Āobs against GT (or HT) should give a rectangular hyperbola with only a particular value of Kass giving a good fit to the experimental data.
Results and discussion
Benzoic acid (BA)
To show the application of the new system in binding studies, particularly the effect of substituents on the binding process, BA was chosen as the parent molecule. The pH titration of BA was performed using the new setup and the pH dependent UV absorption spectra of BA in H2O are presented in Fig. 4a. Critical inspection of the UV spectrum shows evidence of the three absorption systems, 1Lb, CT, and 1La for both the ionised and unionised forms of BA. For the unionised form, the bands are positioned at ∼283, 230 and ∼212 nm respectively. For the ionised form, the bands are positioned at ∼277, 224 and ∼209 nm respectively. The results are in agreement with reported values for COOH substituted benzene.30 Substitution of benzene with polar groups containing unshared electrons shifts the absorption bands to a longer wavelength and also intensifies them due to the interaction of the unshared electrons with the benzene nucleus (π–p conjugation).31 The vibronic components associated with the 1Lb band between ∼290 nm and 260 nm are not cleanly resolved. However, the unionised form shows three peaks; a smaller peak representing the 0–0 origin at ∼283 nm (35336 cm−1), one at ∼280 nm (35714 cm−1, 378 cm−1 component) and a third at ∼273 nm (36630 cm−1, 916 cm−1 component). At pH 7.0 (ionised form), the peaks are weaker, slightly sharper and blue shifted to ∼277 nm and 269.8 nm. The CT bands of the unionised and ionised forms of benzoic acid have ε230 ∼ 11500 and ε224 ∼ 8950 respectively. The 1La bands have ε212 ∼ 6100 and ε209 ∼ 3000 for the unionised and the ionised forms respectively. Good isosbestic points were observed and plotting A274 against pH gave pKa = 4.3 (Fig. 5a), which is in close agreement with reported values.32
 | ||
| Fig. 4 UV and CD spectra of BA (0.001 M) in the free and the bound form at pH 2.0 and pH 7.0 in 1.0 and 0.1 cm (a) pH dependent UV spectra of BA in the free form, (b and c) pH titration spectra of BA/β-CyD complex, UV and ICD respectively (d and e) β-CyD effect on UV and ICD spectra respectively at pH 2.0 (f and g) β-CyD effect on UV and ICD spectra respectively at pH 7.0. | ||
 | ||
| Fig. 5 Titration curves of BA in H2O, in 1.0 and 0.1 cm cell (a) pH titration curve of free BA (b) pH titration curve of BA/β-CyD complex (c) binding curves at pH 2.0 (d) binding curves at pH 7.0 (e) analysis of binding curve showing 1:1 stoichiometry of BA/β-CyD complex (f) possible co-conformation of BA/β-CyD complex at pH 2.0 and pH 7.0. | ||
β-CyD titration of BA at pH 2.0 was performed as described earlier starting from a 0.001 M BA only solution and adding appropriate volumes of the 1:15 (molar ratio) BA/β-CyD solution. BA is an achiral molecule, which does not have a CD spectrum. However, in the chiral environment of β-CyD, CD is induced. UV absorbance and ICD spectra of BA binding to β-CyD at pH 2.0 are presented in Fig. 4d and e.
There is an apparent increase in the absorption intensity of the 1Lb band with a ∼1 nm wavelength red shift upon addition of β-CyD indicating hydrogen bonding between BA–COOH and β-CyD. The CT band shows a decrease in absorption intensity upon addition of β-CyD with no wavelength shift. The vibronic components of the 1Lb band became more defined with increasing β-CyD concentration, which indicates that the ring system of BA is in the nonpolar environment of the β-CyD cavity. The ICD band corresponding to the 1Lb is bisignate with a crossing point from negative to positive at ∼270 nm which lies approximately in the centre of the 1Lb absorption band. The intensity increases with increasing β-CyD concentration to a point where further addition has no effect on the ICD spectra. ICD can be either positive or negative depending on the co-conformation of the binding complex (alignment of the transition moments of BA inside or outside the cavity of β-CyD).33 The bisignate nature of the ICD signal is explained by the presence of two conformers resulting in the alignment of two transition moments with the axis of β-CyD. The major positive ICD signal near 230 nm corresponds to the phenyl chromophore transition aligned slightly parallel to the axis, which is most probably due to restricted co-conformation enforced by hydrogen bonding with the secondary hydroxyl rim. The other transition moment between the ring system and the COOH group is aligned orthogonally to the axis giving rise to negative ICD, and thus at pH 2.0 (unionised form) BA is adopting a slightly tilted lateral co-conformation (Fig. 5f) which gives the bisignate ICD (Fig. 4e).34 These results are in close agreement with structural assignments produced by NMR and crystallography.35,36 The binding constant (K) at pH 2.0 was determined by plotting the value of A274 and ΔA274 against β-CyD concentration (Fig. 5c) and the analysis of the binding curve produced a mean K = 5.63 × 102 M−1.
At pH 7.0, the UV and CD spectra of the BA/β-CyD titration are presented in Fig. 4f and g respectively. The absorption 1Lb band shows a decrease in intensity and there is no wavelength shift with increasing β-CyD concentration due to the loss of hydrogen bonding indicating that BA-COO− does not participate as a hydrogen bond acceptor most probably due to solvation effect at high pH confirming that hydrogen bonding of COOH group is the main force contributing to the stability of the binding complex. Also, the vibrational structure is not as well defined as it is at pH 2.0 suggesting that the ring system is not immersed inside the nonpolar environment of the β-CyD cavity, or the molecule is in and out of the cavity to minimise its net dipole moments by taking a more stable co-conformation. The absorption CT band shows an increased intensity with increasing β-CyD concentration this implies that there is an interaction most probably hydrophobic interaction between the ring system and β-CyD as a result of being in close proximity to the β-CyD cavity. On the other hand, the ICD associated with the 1Lb and CT bands is very weak with the disappearance of bisignate characteristic of the 1Lb band suggesting that the loss of hydrogen bonding gave the molecule more freedom and resulted in the presence of only one conformer, which is not immersed deeply in the β-CyD cavity adopting unstable tilted frontal co-conformation as shown in Fig. 5f. The binding curve produced by plotting the values of ΔA254 against β-CyD concentration (Fig. 5d) produced a very small value of K = 0.53 × 102 M−1 confirming that the ionised form of BA does not form a stable complex with β-CyD. Analysis of the binding curve (Fig. 5e) show a stoichiometry of 1:1 for BA/β-CyD binding complex. The pH titration of the bound form of BA at 1:15 molar ratio (BA/β-CyD) (Fig. 5b and c) confirms the results obtained previously. As expected, the ICD collapses going from pH 2.0 to pH 7.0, however, plotting the values of A274 and ΔA274, against pH (Fig. 5b) produces a mean pKa = 5.0, which is significantly higher than the pKa of the free form of BA, this increase in pKa value, or decrease in BA acidity suggests that the CO2H resonance is disrupted by hydrogen bonding with β-CyD; also the co-conformation or steric effect on entropy makes CO2H less exposed to the solvent molecules resulting in the decreased acidity.
m-Chlorobenzoic acid (m-Cl-BA)
The pH-dependent UV-absorption spectra of free m-Cl-BA with two different pathlengths (0.1, 1.0 cm) are shown in Fig. 6a. The 1Lb, CT, and 1La bands show a red shift compared to BA with the biggest shift of ∼8 and ∼7 nm in the 1Lb band at pH 2.0 and pH 7.0 respectively. The CT band is positioned at 232 nm at pH 2.0, and 229 nm at pH 7.0, however, the 1La band is positioned at ∼217 nm at both pHs. A plot of A283 against pH (Fig. 7a) gave a value of pKa = 3.8. This value is considerably smaller than the pKa values of BA (pKa = 4.3), which is due to the effect of Cl at the meta position on the CO2− group making it less susceptible to the solvent as a result of charge dispersal and a change in electron density distribution in the CO2H group resulting in easier loss of H upon titration. In general, substitution of benzene by auxochromes (nonchromophoric groups, usually containing unshared electrons such as –Cl, –NR, –OH), has varying effects on the absorption spectra, with the position of the substitution is of great importance to the developed spectra. | ||
| Fig. 6 UV and CD spectra of m-Cl-BA (0.001 M) in the free and the bound form at pH 2.0 and pH 7.0 in 1.0 and 0.1 cm (a) pH dependent UV spectra of m-Cl-BA in the free form, (b and c) pH titration spectra of m-Cl-BA/β-CyD complex, UV and ICD respectively (d and e) β-CyD effect on UV and ICD spectra respectively at pH 2.0 (f and g) β-CyD effect on UV and ICD spectra respectively at pH 7.0. | ||
At pH 2.0, the binding titration of 0.001 M m-Cl-BA with a solution of m-Cl-BA/β-CyD (1:15) was performed in the same way using 1.0 and 0.1 cm cells and the resulting UV and ICD spectra are shown in Fig. 6d and e. The 1Lb absorption is slightly decreased and red shifted by ∼2 nm with increasing β-CyD concentration. The vibrational structure becomes more defined with increasing β-CyD concentration, which can be seen in the vibronic components at 292 and 285 nm as they became sharper upon increasing β-CyD concentration than it is the case with BA. This indicates that the ring system is immersed deeper inside the nonpolar cavity of β-CyD. The ICD shows optical activity very similar to benzoic acid, it is bisignate with a crossing point from negative to positive at 268 nm. However, the ICD of the 1Lb band is significantly more intense than that of BA, indicating the effect of Cl on the charge distribution and the alignment of the transition moments within the β-CyD cavity. On the other hand, the addition of β-CyD does not cause any wavelength shift of the CT or the 1La band. Below 210 nm, the UV and CD spectra are dominated by the 1Ba transition. However, the spectra are distorted due to the high absorbance value, thus measurements of this transition under these conditions are not reliable. The binding curves of m-Cl-BA/β-CyD interaction (Fig. 7c) are produced by plotting the values of A233 and ΔA233 against β-CyD concentration. The mean value obtained was K = 4.03 × 103 M−1 which is significantly higher than that of BA (∼10 fold increase) as result of m-Cl-substitution.
 | ||
| Fig. 7 Titration curves of m-chlorobenzoic acid in H2O, in 1.0 and 0.1 cm cell (a) pH titration curve of free m-chlorobenzoic acid (b) pH titration curve of m-chlorobenzoic acid/β-CyD complex (c) binding curves at pH 2.0 (d) binding curves at pH 7.0 (e) analysis of binding curve showing 1:1 stoichiometry of m-Cl-BA/β-CyD complex (f) possible co-conformation of m-Cl-BA/β-CyD complex at pH 2.0 and pH 7.0. | ||
At pH 7.0, the effect of β-CyD titration on the UV and ICD spectra of the ionised form of m-Cl-BA are shown in Fig. 7f and g respectively. There is no significant wavelength shift at this pH compared to the free form of m-Cl-BA and the spectra are less intense, however, contrary to the non-ionised form, the intensity of the absorption bands increases with increasing β-CyD concentration. Also, the 1Lb vibronic structure is well defined and shows three peaks at ∼285, 277 and ∼268 nm, indicating that the ring system is inside the cavity and being stabilised mainly by Cl and hydrophobic interactions. Unlike the ionised form of BA, the ionised form of m-Cl-BA exhibits a bisignate ICD signal with a crossover point at ∼273 nm and two vibronic components at ∼285 and ∼277 nm, which indicate the effect of m-Cl on the conformational stability of the binding complex, forcing the molecule to adopt two conformers at different energy states. The binding curves of A285 and ΔA285 against β-CyD concentration produced a value of K = 1.29 × 102 (Fig. 7d). The relatively small value of K indicates that the main contributor to binding stability is H-bonding and therefore, the ionised form of m-Cl-BA complex is less stable. The effect of pH change on the UV absorption and ICD spectra of the bound form of m-Cl-BA is presented in Fig. 6b and c. Good isosbestic points are observed indicating a two states system (unionised/ionised). The ICD intensity decreases going from pH 2.0 to pH 7.0 with isosbestic points at ∼258 and ∼214 nm. The ICD at these wavelengths is equal for both ionised and unionised form suggesting the same alignment of these transitions with β-CyD at both the ionised and non-ionised forms.
 | ||
| Fig. 8 β-CyD titration of p-Cl-BA (0.001 M) at pH 2.0 and pH 7.0 in 1.0 and 0.1 cm (a) effect of β-CyD on UV spectra (b) ICD spectra (c) binding curves at pH 2.0 (d) binding curves at pH 7.0 (e) possible co-conformation of p-Cl-BA/β-CyD complex at pH 2.0 and pH 7.0. | ||
Plotting the values of absorbance and ICD at A283, and ΔA283 against pH (Fig. 7b) gives a mean value of pKa = 4.92, which is significantly higher than that of the free acid (pKa = 3.8). This decrease in acidity upon inclusion in β-CyD signifies the effect of β-CyD interaction with COOH and Cl− on the charge distribution around the H of the carboxyl group making it less susceptible to the solvent resulting in a higher pKa value. Analysis of the binding curve (Fig. 7e) confirms a 1:1 stoichiometry of the m-Cl-BA/β-CyD complex. The results confirm that the Cl substitution at the meta position has made a significant contribution to binding stability at both pHs.
p-Chlorobenzoic acid (p-Cl-BA)
The effect of substitution position can be emphasised by using p-Cl-BA to show the effect of Cl in the para position on binding to β-CyD. The pH dependent absorption spectra of the free form of p-Cl-BA (results not shown) shows in comparison to BA, no wavelength shift in the 1Lb (282 nm), however, the 1La (214 nm) band is red shifted by ∼4 nm at both pH 2.0 and pH 7.0. The CT band shows a significant red shift of ∼12 nm at both pHs indicating the inductive effect of p-Cl on the ring system. Plotting A274 and A217 nm (results not shown) against pH gave a value of pKa = 4.2, which is slightly lower than pKa of BA and significantly higher than pKa of m-Cl-BA as a result of field effect of Cl− on the CO2H group. The absorbance and ICD spectra of p-Cl-BA titration with β-CyD at pH 2.0 are shown in Fig. 8a and b respectively. The UV spectra exhibit a decrease in intensity with the addition of β-CyD accompanied by ∼1 nm red shift of both the 1Lb and CT bands. The red shift and change in intensity of the 1Lb band suggests hydrogen bonding of the carboxyl group; however the red shift of the CT band indicates that a lower energy transition has occurred as a result of binding to β-CyD. There is no significant change in either intensity or position of the 1La band located at 214 nm. The ICD of the CT transition matches the absorption wavelength and its intensity increases with increasing β-CyD concentration. However, the ICD of the p-Cl-BA 1Lb band at ∼283 nm is more complex than its counterpart in BA. It is again bisignate with a crossover point at ∼280.5 nm, nevertheless, the 1Lb ICD is considerably weaker than the BA counterpart due to the different angle of the transition moment alignment inside the β-CyD cavity. The CT absorption and ICD bands increase in intensity with increasing β-CyD concentration. However, the ICD signal at the lower wavelength range is also bisignate, going from positive to negative ICD with a crossing point at ∼214 nm and a negative maximum at ∼208 nm most probably associated with steric effect resulting from p-Cl forcing the molecule into a negative co-conformation with increase in intensity and a shift towards higher energy (shorter wavelength) upon increase of β-CyD concentration. The binding curves produced by plotting the values of ΔA242 and ΔA207 against β-CyD concentration are shown in Fig. 8c. The mean value of K = 2.47 × 102 M−1 indicates a relatively low binding at pH 2.0 in comparison to m-Cl-BA, confirming the significant effect of substituent position on the stability of the binding complex.At pH 7.0, where p-Cl-BA is completely ionised; the resulting absorption and CD spectra are similar to those at pH 2.0 (results not shown) As at pH 2.0, the 1Lb absorbance decreases in intensity and exhibits a blue shift of 1 nm with increasing β-CyD concentration. At pH 7.0, more of the 1Lb vibronic structure of p-Cl-BA bound to β-CyD is revealed due to the immersion of the ring system into the β-CyD cavity. Collectively, the intensity of the ICD at pH 7.0 is almost as half as that at pH 2.0 and the negative ICD band at ∼208 nm is not clearly defined. ICD is angle dependent and the loss of hydrogen bonding in the CO2− gave more freedom to the molecule in the cavity, which resulted in a change of the angle of co-conformation with β-CyD and a lower intensity ICD.
Plotting A270 and ΔA237 against β-CyD concentration (Fig. 8d) gave mean K = 2.11 × 102 M−1, which is slightly less than the K value at pH 2.0 (K = 2.47 × 102 M−1). In the same way, the pH titration of bound form of p-Cl-BA was performed and plotting absorbance and ICD values against pH gave pKa = 3.83, which is less than the pKa of the free form of p-Cl-BA. The increased acidity of p-Cl-BA in the bound form is mostly due to immersion of the molecule in the β-CyD cavity and the polarized local environment around the COOH, which made it more susceptible to the solvent, in addition to the field effect of p-Cl that might have become closer to the CO2H in the environment of β-CyD due to changes in the molecule conformation resulting in stabilising the COO− and the easier loss of the H ion. According to the resulting ICD, the p-Cl-BA molecule seems to assume a tilted frontal co-conformation with the Cl going in first and deep into the cavity (Fig. 8e). This assumption is in close agreement with results obtained from NMR analysis, which state that the center of the aromatic ring of the drug molecule was considered to be located in the cavity 1.2 Å inside from the H-5 plane of β-cyclodextrin.37 However, the bisignate nature of the ICD suggests that the molecule attempts to minimise its net dipole moment in the nonpolar environment of the β-CyD (low dielectric) by adopting different conformers resulting in the overlapping of a shorter wavelength (∼208 nm) negative ICD with a positive ICD and another negative but small ICD at higher wavelengths due to the angle of alignment of these transition moments within the β-CyD environment. The data suggests that although p-Cl-BA has less binding affinity than both BA and m-Cl-BA, the conformational equilibrium of p-Cl-BA is more sensitive to the β-CyD environment than BA or m-Cl-BA. This confirms that the location of the substituents can cause a significant difference to the binding complex.
Phenylacetic acid (PAA)
PAA shows the effect of introducing a saturated methylene group between the two chromophores (CO2H–ring system) on binding affinity. Due to solubility constraints, the titration of PAA was performed in a 2.0 cm cell for the wavelength range (300–230 nm) and a 1.0 cm cell for the range 260–190 nm. The pH titration (spectra not shown) showed good isosbestic points, which validate the reliability of the titration and the existence of a two state system (unionised and ionised). The vibronic structure is apparent in the symmetry forbidden weak 1Lb band, with the 0–0 component at ∼267 nm and at ∼268 nm for the unionised and ionised form respectively. In the far UV region, the 1La transition band of the unionised and ionised form is at ∼206 nm and 207 nm respectively, both with ε = ∼8100. There are pronounced shoulders at ∼215 nm (ε = 3600) and 218 nm (ε = 4500) for the unionised and ionised form respectively. This indicates another transition due to intramolecular electron/charge transfer to the π-system of the aromatic ring, which can be confirmed by the notably increased intensity and red shift of the ionised form. Plotting various absorbance values against pH gave a mean pKa = 4.35, which is in agreement with literature values.38The titration of PAA with β-CyD at pH 2.0 showed no significant change in the far UV region (results not shown). Then again, in the 300–230 nm range, the 1Lb UV band (Fig. 9a) is effectively unchanged with the addition of β-CyD and there is no evidence of fine structure ICD associated with the 1Lb transition (Fig. 9b) indicating that the ring system is not in the nonpolar environment of β-CyD. However, there is a slight decrease in absorbance running from 260 nm to 235 nm accompanied by a weak positive ICD as a result of the inclusion of PAA in the dissymmetric environment of β-CyD. The relatively small change in the ICD at 260–230 region implies that any binding is most probably due to weak hydrogen bonding between the CO2H moiety and β-CyD outer rim. The binding curve obtained by plotting ΔA240 against β-CyD concentration (Fig. 9c) produced a value of K = 1.15 × 102 M−1, which is very low and indicative of non specific binding in agreement with the low ICD results. At pH 7.0, phenylacetic acid is ionised, the β-CyD titration showed no significant effect on the UV spectra and there is effectively no evidence of ICD (spectra not shown). However, the absorbance intensity of all transitions shows very slight increase with increasing β-CyD concentration opposite to the case at pH 2.0. The results indicate that there is apparently no PAA/β-CyD binding at pH 7.0 confirming that at pH 2.0, the weak binding is due to weak forces mainly hydrogen bonding. So far, there are no reports that show any binding of the ionised form of PAA to β-CyD. The binding of PAA to β-CyD indicates that introducing a saturated methylene group between the two chromophores may cause a loss of flexibility resulting in a poor fit within the cavity, which significantly reduces the binding affinity.
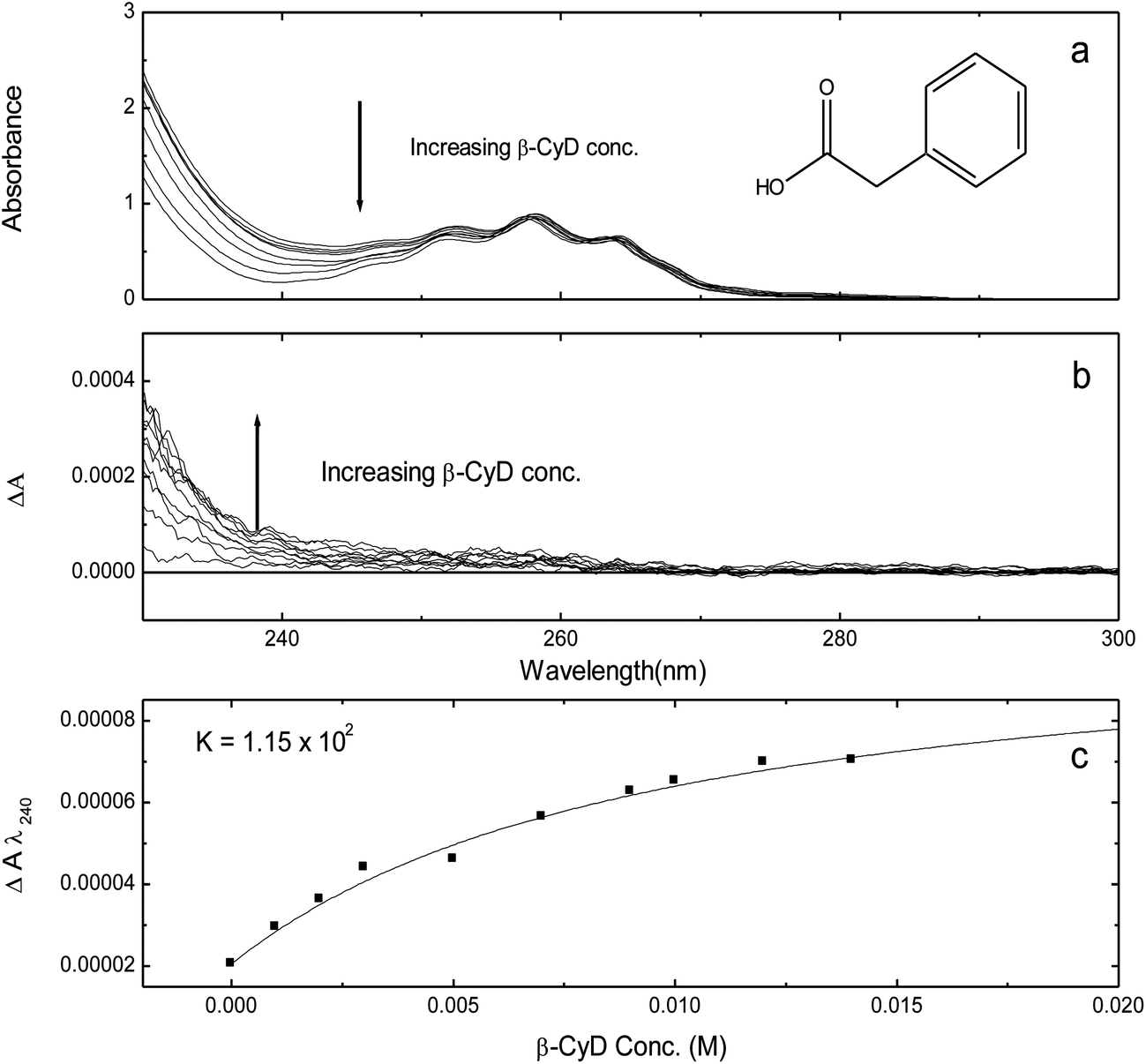 | ||
| Fig. 9 β-CyD titration of PPA (0.001 M), pH 2.0, in H2O, 2.0 cm. | ||
The pH titration of PAA/β-CyD at a molar ratio of 1:14 showed no significant ICD as a result of phenylacetic acid/β-CyD interaction (results are not shown). Nevertheless, plotting A260 and A235 against pH gave a mean pKa = 4.6, which is slightly higher than the pKa for the free form of PAA (pKa = 4.35). The slight increase in the pKa value indicates that PAA is slightly less acidic in the presence of β-CyD. The change in pKa again confirms that there is interaction between PAA–CO2H group and β-CyD, albeit weak and only at acid pH. This weak interaction made the CO2H slightly resistant to the solvent and decreased the acidity by ∼0.25 pH units.
Similarly, the effect of substituents of other compounds on binding interaction was studied using the new system.26 These compounds included S-(+)ibuprofen (S-Ibu) as the pharmacologically active component of the racemic mixture, aspirin (acetyl salicylic acid, ASA) and salicylic acid (SA), which are important type of drugs (NSAIDs) and are one of the most commonly used drugs that are sold over the counter. The results for all compounds are summarised in Table 1. In the case of S-Ibu, which is a chiral molecule, the introduction of an isobutyl group has shifted all transitions to a longer wavelength (a red-shift) with the phenyl related transition ε264 = ∼380 (spectra not shown). At pH 7.0, the change in the UV absorbance and CD of S-Ibu with increasing β-CyD concentration suggests an interaction between both compounds and thus a concomitant complex formation at this pH. The 1Lb band shows two isosbestic points at 280 nm and 272.6 nm, which indicates a 1:1 stoichiometry of S-Ibu/β-CyD complex.39 At pH 7.0, the ionised form of S-Ibu is not restricted by hydrogen bonding to the β-CyD rim and the molecule has more freedom to immerse deeper into the cavity. The CD spectra are negative in sign and they increase in intensity with increasing β-CyD concentration. The increase or the ICD resulting from interaction with β-CyD is negative and more intense suggesting a frontal co-conformational inclusion. The presence of the isobutyl group may be a cause of deeper penetration into the cavity, because the affinity of the isobutyl group for the β-CyD cavity is favorable since the inner walls of β-CyD are considerably hydrophobic, this has been substantiated in literature by interaction energy analysis and force field calculations.40,41 Also, the vibrational fine structure does not exhibit any change suggesting that the ring is far from the non-polar part of the cavity which leads to conclude that the isobutyl group is inside the cavity near the nonpolar end, whereas it is highly unlikely for the polar CO2H group to be near the nonpolar part of the cavity. The K value of the ionised S-Ibu is considerably higher than that of the unionised form (Table 1), implying that hydrogen bonding restricts the molecule flexibility and prevents it from deeper penetration resulting in a lower K value, which signifies the effect of isobutyl group on the binding interaction. This can be confirmed by the increased acidity upon complexation (Table 1) and a co-conformation, which has led to stabilisation of the ionised form and easier loss of the hydrogen ion. On the other hand, ASA contains a substituted phenyl ester group which is a nucleophilic center susceptible to hydrolysis and other acyl transferring reactions.42 It can undergo acid-catalyzed ester hydrolysis, as well as a base-promoted ester hydrolysis and the formation of salicylic acid as a hydrolysis product. The results showed evidence of ASA acetyl hydrolysis in the 1.0 cm pathlength range (320–230 nm) as a peak was appearing at ∼300 nm with time and increasing pH as a result of salicylic acid presence in the solution. Binding titration at pH 7.0, the results (not shown) showed no ICD upon complexation with β-CyD indicating that the ionised form of ASA adopts no co-conformation which could induce optical activity suggesting that the ionized ASA does not form complexes with the β-CyD.43 However, at pH 2.0, in comparison to the unbound or free ASA, again the UV spectra show a small red shift of the 1Lb band of ∼1 nm and increased intensity with increasing β-CyD concentration, as evidence of hydrogen bonding. At pH 2.0, the unionised form of ASA showed a positive ICD and the bisignate nature has disappeared signifying the effect of the acetyl group on the conformation of ASA in the β-CyD environment. The major ICD signal is near 230 nm, which correlates with the CT band, it corresponds to the phenyl chromophore transitions and is most probably due to restricted co-conformation enforced by the acetyl group and the formation of hydrogen bond with the secondary hydroxyl rim, and thus, adopting a tilted lateral co-conformation with the phenyl ring slightly immersed inside the cavity, which gives the positive ICD. The binding constant at pH 2.0 was determined; K = 1.49 × 102 M−1, which is in close agreement with reported values.44 The β-CyD titration gives two isosbestic points in the UV spectra near 300 and 260 nm indicative of a two state free/bound 1:1 stoichiometry of the ASA/β-CyD inclusion complex. The pH titration of the bound form of ASA gave a pKa = 4.14, which is significantly higher than the pH of the free form (pKa = 3.6). This suggests that the CO2H resonance is disrupted by hydrogen bonding with β-CyD; also the co-conformation or steric effect on entropy makes the hydrogen less exposed to the solvent molecules resulting in the decreased acidity. In addition, the UV band at ∼300 nm that was apparent in the pH titration of the free form has disappeared or significantly decreased indicating a decelerating effect of β-CyD binding on hydrolysis. This is more likely as a result of shielding of the ester group due to steric hindrance by β-CyD.
| Compound | Substituent | K (× 102 M−1) | pKa | KCyD:HA/KCyD:A− | ||
|---|---|---|---|---|---|---|
| Ionised | Non-ionised | Free form | Bound form | |||
| BA |  |
0.53 | 5.63 | 4.30 | 5.00 | 10.62 |
| m-Cl-BA |  |
1.29 | 40.03 | 3.80 | 4.90 | 31.03 |
| p-Cl-BA |  |
2.11 | 2.47 | 4.20 | 3.83 | 1.17 |
| PAA |  |
N/A | 1.15 | 4.35 | 4.60 | N/A |
| (+)-(S)-Ibu |  |
27.37 | 13.17 | 5.20 | 4.60 | 0.48 |
| ASA |  |
N/A | 1.49 | 3.60 | 4.14 | N/A |
| SA |  |
1.05 | 4.14 | 2.80 | 3.40 | 3.94 |
In the same way, salicylic acid (SA) pH titration gave a mean pKa = 2.8, which is in agreement with literature values.45,46 The protonated form of SA exhibited the three transitions 1Lb, CT and 1La at ∼303 nm, 238 nm and ∼218 nm respectively. However, at pH 7.0, the ionised form three transitions were at ∼300 nm, ∼230 nm and ∼205 nm. Titration of SA with β-CyD at pH 7.0 showed no induced CD, this suggests that SA− ion does not bind to β-CyD, or most probably the structural co-conformation does not induce any CD, however, it was reported in literature that the salicylate ion, albeit weakly binding (Table 1), it does bind to β-CyD.47
Therefore, it is reasonable to ascribe binding of the ionised form to weak forces e.g. hydrophobic interaction. Moreover, when the molecule is in the ionised form it is acting as both hydrogen bond donor and acceptor at the same time due to the loss of intramolecular hydrogen bonding between OH group and CO2H group upon ionisation leaving the ortho-OH group as a possible hydrogen bond donor and COO− as hydrogen bond acceptor, which makes it unsusceptible to the environment of β-CyD, therefore the 1Lb band exhibits no shift and no ICD observed. The titration of SA with β-CyD at pH 2.0 showed a red shift of the 1Lb absorption band by ∼5.0 nm, indicating hydrogen bonding.
Again, the ICD of the 1Lb band is bisignate with a crossover point from negative to positive at ∼270 nm similar to that of BA. The CT and 1La transitions have positive ICD at ∼240 and ∼205 nm respectively. The bisignate feature most probably suggests two different conformers of SA at the corresponding wavelengths with a slightly tilted frontal co-conformation of SA/β-CyD inclusion complex, where the 1Lb transition is orthogonally aligned in the β-CyD cavity giving a small negative ICD and the CT and 1La transitions are in a slightly tilted parallel alignment giving a small positive ICD. The results indicate that the H on the CO2H group on SA is again the main contributor, which interacts with the nucleophilic OH of β-CyD. The pH titration of the bound form of SA gave a mean value of pKa = 3.4, suggesting loss of the resonance effect due to interaction with β-CyD. In addition, conformational change may cause the loss of intramolecular hydrogen bonding in SA, which contribute to the low acidity of SA making it harder to lose the H upon NaOH addition and consequently decreased acidity. In comparison to the bound form of BA, the increased pKa of the bound form of SA as well as the binding constant (K = 4.14 × 10 2 ± 16 M−1, which is in close agreement with reported values44) are slightly less indicating the effect of OH group on binding stability.
The binding constants as an indication of the stability of the inclusion complexes of these compounds in the protonated form are in this order, m-Cl-BA > S-Ibu > BA > SA > p-Cl-BA > ASA > PAA. Table 1 show the different K values for these compounds and the ratios between KCD:HA/KCD:A− for each compound, which is characteristic of the guest/β-CyD complex and must then be constant. The CO2H group when involved in an intramolecular charge transfer, makes a substantial contribution to the ground state,30 but their predominant contributions are to the excited state, resulting in a bathochromic shift accompanied by hyperchromism. Moreover, according to the extensive studies by Doub and Vandenbelt48 on the effects of substituents on the 1La band of benzene, they found that the ratio of the wavelengths of the 1Lb band and the 1La band is about 1.25 for most mono-substituted benzenes including benzoic acid. The results show that the ratio between the 1Lb band and the 1La band assigned here is about 1.25, 1.25 and 1.29 for PAA, S-Ibu and BA respectively. The ratio between the 1Lb band and the CT band is between ∼1.15 and 1.26 for all compounds.
The values of these ratios for all the compounds at pH 2.0 based on λmax (Table 2) are in close agreement with the assumption that these bands are due to intramolecular charge transfer.
| Compound | 1Lb (nm) | CT (nm) | 1La (nm) | 1Lb/CT | 1Lb/1La |
|---|---|---|---|---|---|
| BA | 274 | 230 | 212 | 1.19 | 1.29 |
| m-Cl-BA | 283 | 232 | 217 | 1.21 | 1.30 |
| p-Cl-BA | 276 | 240 | 214 | 1.15 | 1.28 |
| PAA | 258 | 218 | 206 | 1.18 | 1.25 |
| S-Ibu | 265 | 225 | 212 | 1.18 | 1.25 |
| ASA | 278 | 230 | 211 | 1.20 | 1.31 |
| SA | 300 | 238 | 218 | 1.26 | 1.37 |
Conclusion
An important advantage of the multi-cell low volume device for the simultaneous measurement of absorption and circular dichroism, is that it does not affect the essential requirements for the UV/CD technique; therefore, it can be used in all UV/CD applications. It proved to be an efficient and a reliable apparatus, which can be used routinely in pH and binding studies (the results obtained using this system are in agreement with reported values). Furthermore, it is a versatile, compact, and low-volume apparatus that makes it possible to simultaneously use three different pathlengths plus a reference cell, without the need for manual transfer of solution and the consequent decrease of measurement solution volume, which can have a significant effect on results outcome. Additionally, low volume (≤2 ml) is used, and it can even be lower volume by using specially manufactured smaller flask, which is particularly important when the amount of available samples is limited. It is an efficient mixing apparatus in pH and binding titration particularly with small volumes, also, the variable speed pump is very useful when gentle mixing is required as it is the case in protein studies to avoid mechanical aggregation. In general, the analysis time is significantly reduced to ≤1/4 of total analysis time used in a one-cell system. Moreover, this device can readily be automated for routine high throughput analysis. However, the only disadvantage was that after rinsing the cells, they were not completely empty. The amount of water or rinse solution left in the cell was ≤100 μl, which was readily accounted for in the calculation. This problem was solved by rinsing the cells with ethanol solution and then attaching the cell tubing to a vacuum to make sure that the cells are completely empty.The charge on the guest molecule, its structure and substituents are of great importance in regulating the stability of the guest/β-CyD complexes; hence the bioavailability of drugs. What is apparently generally neglected is that the CO2H group is more likely to participate in H-bonded structures than CO2− particularly if it is an H-donor. Changes in the UV and/or CD spectra upon addition of β-CyD have provided evidence of the significant effects of substituents on binding interaction. Also, the change in the guest environment is evident in the vibronic structure of the molecules upon complexation. The change in spectroscopic variables (UV, CD and ICD) have provided good quantitative measures for substituent effects on pKa, binding constant and stoichiometry. Disubstituted benzenes can be the subject of a future work as another application of the new device. Drug binding to Cyclodextrin is not well understood, however, the results undoubtedly helped to assign possible structural co-conformations for the drug/β-CyD inclusion complex increasing our understanding of drug/cyclodextrin interactions.
Acknowledgements
Many thanks Dr Dhia El-Hag, molecular biophysics group at King's College London and Applied Photo Physics (APL) Ltd. for their support.Notes and references
- J. M. Lehn, Supramolecular Chemistry, VCH, Weinheim, 1995 Search PubMed.
- M. Filippa, M. I. Sancho and E. Gasull, J. Pharm. Biomed. Anal., 2008, 48, 969–973 CrossRef CAS PubMed.
- M. Abdel-Tawab, H. Zettl and M. Schubert-Zsilavecz, Curr. Med. Chem., 2009, 16, 2042–2063 CrossRef CAS.
- G. Schmid, Trends Biotechnol., 1989, 7, 244–248 CrossRef CAS.
- M. Roux, B. Perly and F. Djedaini-Pilard, Eur. Biophys. J., 2007, 36, 861–867 CrossRef CAS PubMed.
- T. Loftsson and D. Duchene, Int. J. Pharm., 2007, 329, 1–11 CrossRef CAS PubMed.
- G. Tiwari, R. Tiwari and A. K. Rai, J. Pharm. BioAllied Sci., 2010, 2, 72–79 CrossRef CAS PubMed.
- R. A. Rajewski and V. J. Stella, J. Pharm. Sci., 1996, 85, 1142–1169 CrossRef CAS PubMed.
- J. Szejtli and T. Osa, in Comprehensive Supramolecular Chemistry, ed. J. M. Lehn, Pergamon Press, Oxford, UK, 1996, vol. 3 Search PubMed.
- F. L. Aachmann, D. E. Otzen, K. L. Larsen and R. Wimmer, Protein Eng., 2003, 16, 905–912 CrossRef CAS PubMed.
- J. Geczy, J. Bruhwyler, J. Scuvee-Moreau, V. Seutin, H. Masset, J. C. Van Heugen, A. Dresse, C. Lejeune, E. Decamp, L. Szente, J. Szejtli and J. F. Liegeois, Psychopharmacology, 2000, 151, 328–334 CrossRef CAS.
- E. Junquera, M. Martin-Pastor and E. Aicart, J. Org. Chem., 1998, 63, 4349–4358 CrossRef CAS.
- P. Mura, S. Furlanetto, M. Cirri, F. Maestrelli, G. Corti and S. Pinzauti, J. Pharm. Biomed. Anal., 2005, 37, 987–994 CrossRef CAS PubMed.
- Y. Nievergelt, Analyst, 1994, 119, 145–151 RSC.
- N. Sadlej-Sosnowska, L. Gasinski and W. P. Oziminski, Pol. J. Chem., 2003, 77, 1039–1048 CAS.
- K. A. Connors, in Binding Constants, The Measurement of Molecular Complex Stability, ed. K. A. Connors, Wiley-Interscience, New York, 1987 Search PubMed.
- C. D. Tran, M. S. Baptista and T. Tomooka, Langmuir, 1998, 14, 6886–6892 CrossRef CAS.
- Y. Nievergelt, Analyst, 1994, 119, 145–151 RSC.
- Y. Ikeda, F. Hirayama, H. Arima, K. Uekama, Y. Yoshitake and K. Harano, J. Pharm. Sci., 2004, 93, 1659–1671 CrossRef CAS PubMed.
- M. Dockal, D. C. Carter and F. Ruker, J. Biol. Chem., 1999, 274, 29303–29310 CrossRef CAS PubMed.
- G. A. Ascoli, E. Domenici and C. Bertucci, Chirality, 2006, 18, 667–679 CrossRef CAS PubMed.
- O. McConnell, Y. He, L. Nogle and A. Sarkahian, Chirality, 2007, 19, 716–730 CrossRef CAS PubMed.
- J. Frelek, P. Kowalska, M. Masnyk, A. Kazimierski, A. Korda, M. Woznica, M. Chmielewski and F. Furche, Chemistry, 2007, 13, 6732–6744 CrossRef CAS PubMed.
- J. R. Platt, J. Chem. Phys., 1949, 17, 484–495 CrossRef CAS PubMed.
- A. F. Drake, in Protein–Ligand Interaction: Structure and Spectroscopy Practical Approach, Oxford University Press, Oxford, 2001, pp. 123–167 Search PubMed.
- A. Aboel Dahab and D. El-Hag, Anal. Bioanal. Chem., 2012, 404, 1839–1850 CrossRef CAS PubMed.
- A. Aboel Dahab, D. El-Hag and A. F. Drake, Anal. Methods, 2010, 2, 929–935 RSC.
- H. C. Mahler, R. Muller, W. Friess, A. Delille and S. Matheus, Eur. J. Pharm. Biopharm., 2005, 59, 407–417 CrossRef CAS PubMed.
- S. K. Rai, M. Sharma and M. Tiwari, Bioorg. Med. Chem., 2008, 16, 7302–7310 CrossRef CAS PubMed.
- J. B. Lambert, H. F. Shurvell, D. A. Lightner and R. G. Cooks, in Organic Structural Spectroscopy, ed. J. Challice, Prentice Hall, New Jersey, 2001, pp. 274–303 Search PubMed.
- K. Bowden and E. A. Braude, J. Chem. Soc., 1952, 1068–1077 RSC.
- F. H. Clarke, J. Pharm. Sci., 1984, 73, 226–230 CrossRef CAS.
- K. Harata and H. Uedaira, Bull. Chem. Soc. Jpn., 1975, 48, 375 CrossRef CAS.
- H. Bakirci, X. Zhang and W. M. Nau, J. Org. Chem., 2005, 70, 39–46 CrossRef CAS PubMed.
- D. Salvatierra, C. Jaime, A. Virgili and F. Sanchez-Ferrando, J. Org. Chem., 1996, 61, 9578–9581 CrossRef CAS.
- T. Aree and N. Chaichit, Carbohydr. Res., 2003, 338, 439–446 CrossRef CAS.
- C. S. Lu, C. J. Hu, Y. Yu and Q. J. Meng, Chem. Pharm. Bull., 2000, 48, 56–59 CrossRef CAS.
- A. F. McDonagh, A. Phimister, S. E. Boiadjiev and D. A. Lightner, Tetrahedron Lett., 1999, 40, 8515–8518 CrossRef CAS.
- K. A. Connors, Binding Constants: The Measurement of Molecular Complex Stability, John Wiley & Sons, New York, 1987 Search PubMed.
- M. Fujisawa, T. Yasukuni, H. Ikeda, M. Yukawa, H. Aki and T. Kimura, J. Appl. Solution Chem. Model., 2012, 1, 132–138 CAS.
- B. Mayer, X. Zhang, W. M. Nau and G. Marconi, J. Am. Chem. Soc., 2001, 123, 5240–5248 CrossRef CAS PubMed.
- R. O. Williams, 3rd and J. Liu, Eur. J. Pharm. Biopharm., 1999, 47, 145–152 CrossRef.
- T. Loftsson and M. E. Brewster, J. Pharm. Sci., 1996, 85, 1017–1025 CrossRef CAS PubMed.
- E. Junquera, D. Ruiz and E. Aicart, J. Colloid Interface Sci., 1999, 216, 154–160 CrossRef CAS PubMed.
- T. Gravestock, K. Box, J. Comer, E. Frake, S. Judge and R. Ruiz, Anal. Methods, 2011, 3, 560–567 RSC.
- G. A. Ibanez, G. Labadie, G. M. Escandar and A. C. Olivieri, J. Mol. Struct., 2003, 645, 61–68 CrossRef CAS.
- E. Junquera, L. Pena and E. Aicart, J. Pharm. Sci., 1998, 87, 86–90 CrossRef CAS PubMed.
- L. Doub and J. M. Vandenbelt, J. Am. Chem. Soc., 1949, 71, 2414–2420 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |