
Synthetic oligodeoxynucleotide purification by capping failure sequences with a methacrylamide phosphoramidite followed by polymerization†
Abstract
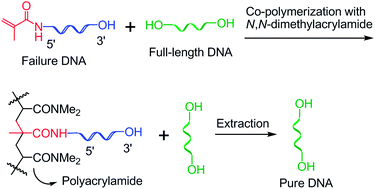
Oligodeoxynucleotide (ODN) purification was achieved by capping failure sequences with a polymerizable methacrylamide phosphoramidite during automated synthesis, polymerizing the failure sequences into an acrylamide gel after cleavage and deprotection, and extraction of full-length sequences with water. The details regarding the technology including the capping efficiency of four polymerizable phosphoramidites, optimal capping time, diffusion speeds of ODN from gels with different cross-linking ratios to solution, and the efficiency of ODN extraction from gel were investigated. In addition, the technology was tested for purification of a long sequence and purification on larger scales. We also found that polymerization of failure sequences in a centrifuge tube in air did not affect purification results. Finally, we provided additional evidence that ODNs are stable under radical polymerization conditions by complete digestion of ODN followed by reversed-phase HPLC analysis of nucleosides.
 Please wait while we load your content...
Please wait while we load your content...

