
Carbon-coated single-crystalline LiFePO4 nanocomposites for high-power Li-ion batteries: the impact of minimization of the precursor particle size
Abstract
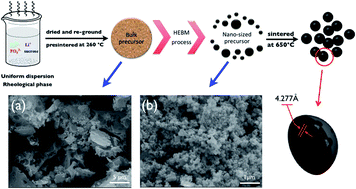
In this work, a high-energy ball mill technique is designed to deal with bulk precursors and achieve particle size minimization. A large amount of the nano-sized precursor is achieved in a narrow particle size distribution of ca. 95 nm. We confirm that the dimensional size of the precursor has a significant influence on the final LiFePO4 particle size and that small grains of the precursor probably form single-crystalline nanoparticles during the calcination process. After carbothermal reduction, the carbon-coated single-crystalline LiFePO4 nanocomposites (nano-CS–LFP) are easily synthesized. Benefiting from the decreasing particle size, the specific surface area of nano-CS–LFP is up to 48.0 m2 g−1, which implies a higher interfacial contact area between the active particles and the electrolyte, as well as an increase in its capacitance capability. Besides, cyclic voltammetry curves of nano-CS–LFP reveal a better capability of reversible reactivity and a lower polarization. Galvanostatic charge–discharge results exhibit excellent rate performance with a discharge capacity of ca. 100 mA h g−1 at 10 °C and a stable cycling property with a capacity retention of ca. 90% after 1000 cycles. In addition, the rapid charge–discharge test over 60 seconds indicates an excellent pulse performance with a high current in a short time period. The combination of the merits of carbon coating and particle size minimization is responsible for the above improvements. Finally, this facile preparation strategy is favorable for the industrial production of economical LiFePO4 materials from lab synthesis.
 Please wait while we load your content...
Please wait while we load your content...

