
Branched platinum–acetylide complexes: synthesis, properties, and their aggregation behavior†
Abstract
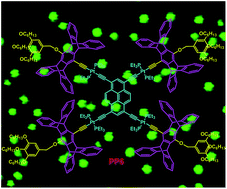
A new branched platinum–acetylide complex PP6 containing pyrene as the main skeleton, pentiptycene units as bridges, and long alkyl chains as branches was successfully synthesized. The structure of PP6 was well characterized by 1H NMR, 31P {1H} NMR, MALDI-TOF-MS spectrometry, and the semiempirical PM6 method. The investigation of the absorption and emission spectra of PP6 and the model complexes revealed that the introduction of iptycene was beneficial to improve the emission efficiency. More importantly, PP6 was aggregated into ordered microspheres driven by the hydrophobic/hydrophilic interactions. The morphologies of the microspheres were investigated by SEM, TEM, LSCM, and EDX. Furthermore, the morphologies and the sizes of the microscale aggregates could be changed by altering the iptycene moiety or the hydrophobic units in PP6.
 Please wait while we load your content...
Please wait while we load your content...

