
Methane hydroxylation using light energy by the combination of thylakoid and methane monooxygenase†
Abstract
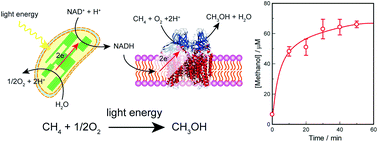
A photoinduced methane hydroxylation system was established by the combination of the photosynthetic system of thylakoid and methane monooxygenase (MMO) from Methylosinus trichosporium OB3b. By using the photosynthetic system, the electrons needed for direct oxidation of methane to methanol can be obtained from water under light irradiation.
 Please wait while we load your content...
Please wait while we load your content...

