
Visible-light photocatalytic mechanism of bisphenol-A on nano-Bi2O3: a combined DFT calculation and experimental study†
Abstract
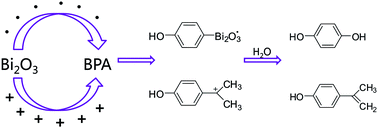
The photocatalytic degradation of bisphenol-A (BPA) on Bi2O3 nanosheets under visible-light irradiation is uncovered and the degradation mechanism is proposed by exploring a reasonable pathway based on theoretical calculations and experiments. For the first time we hypothesized that the photogenerated hole Bi2O3˙+ took part in the early stage of the photocatalytic reaction. With DFT calculations both in the gaseous phase and aqueous phase, five intermediate products were determined: they are hydroquinone, 4-isopropenylphenol, 4-hydroxyacetophenone, methyl p-hydroxybenzoate and p-hydroxybenzoic acid, respectively. To test and verify our calculations, three intermediate products (m/z 151, m/z 133, and m/z 137) were detected by HPLC and LC-MS analyses. By exploring the function of the catalyst Bi2O3, we find that the Bi2O3 catalyst does play an important role in the direct oxidation of holes with bismuth-based semiconductors in the process of visible-light photocatalysis. The catalyst Bi2O3 segments the BPA in the early stage of the photocatalytic reaction and initiates the subsequent degradation reactions, which also serves as a donor of the charges and single electron. Based on the above theoretical calculations and experimental results, we propose a reasonable pathway of the photocatalytic degradation of BPA by Bi2O3. The photogenerated holes Bi2O3˙+ take part in the reaction, which can efficiently reduce the recombination of holes and electrons and thus overcome the deadly shortcoming of photocatalysis. This is critically important in a photocatalytic reaction. Our results will be helpful for understanding the process of visible-light photodegradation.
 Please wait while we load your content...
Please wait while we load your content...

