Silicate anion intercalated cobalt-aluminium hydrotalcite (CoAl-HT-Si): a potential catalyst for alcohol oxidation†
Thangaraj Baskarana,
Raju Kumaravela,
Jayaraj Christopherb and
Ayyamperumal Sakthivel*a
aDepartment of Chemistry, Inorganic Materials and Catalysis Laboratory, University of Delhi, India. E-mail: sakthiveldu@gmail.com; Tel: +91-8527103259
bIndian Oil Corporation Ltd, R&D Centre, Faridabad-121007, India
First published on 6th February 2014
Abstract
Silicate intercalated CoAl hydrotalcite materials were synthesized (CoAl-HT-Si) and systematically characterized. The intercalated hydrotalcite materials lead to the formation of solid solutions of cobalt silicate and cobalt spinel structures, which have higher surface area and pore volume than pure CoAl-HT. The XPS studies revealed the presence of cobalt in divalent and trivalent oxidation states on the surface. The resultant materials were found to be promising catalysts for oxidation of various alcohols.
Introduction
Selective oxidation of alcohols to aldehydes and ketones is a subject of major interest in organic synthesis, as well as an important precursor in fine chemical and pharmaceutical industries.1 Substantial studies on catalytic oxidation of alcohols using noble metals, metal-oxide based catalysts such as Ru,2 Pd,3 Pt,4 Au5 and several other transition metal-containing catalysts6 have been reported. Although transition metal oxide catalysts are known to be effective catalysts for liquid-phase oxidation of many organics,2–6 their applications have been limited owing to their highly toxic nature and noble metal catalysts are highly expensive and scarce. In contrast, heterogeneous systems have the advantages over homogenous systems because the catalysts can be recovered and reused after the reactions.7 Among the various support known, there is a growing interest in the application of hydrotalcite-like compounds (HT), which are composed of positively charged, tri-octahedral hydroxide layers with interlayer anions, and represented by the general formula of [M(II)(1−x)M(III)x(OH)2]x+[An−]x/nmH2O.7–16 These compounds vary in their composition as well as the properties with the substitution of cation in the brucite layer, and the interlayer anions yield a variety of tailor-made materials.17–19 The resultant materials were demonstrated as potential catalysts, catalytic supports, ion-exchangers, adsorbents, flame retardants, corrosion inhibitors and pigments.7–24 It is worth mentioning that CoAl-HT present with interesting magnetic and catalytic properties, however, the catalytic activity mostly arises from the surface concentration of cobalt.25 In this regards, we recently found that layered structure of HT was partially stabilized by interlayer silicate anions species.26 Which we believe can help on connecting hydroxide layers covalently through a porous anionic pillar.17–19,26 Thus, our objective is to introduce silicate anion into the layered CoAl-HT to produce porous layered materials with surface-exposed cobalt species, which might be potential as oxidation catalysts.Here we report the synthesis of silicate anion intercalated cobalt-aluminium hydrotalcite as stable, effective heterogeneous catalysts for the oxidation of alcohols into aldehydes–ketones under mild conditions as green processes. A series of CoAl-HT and silicate anion intercalated samples were prepared according to modified literature and systematically characterized (Scheme 1).
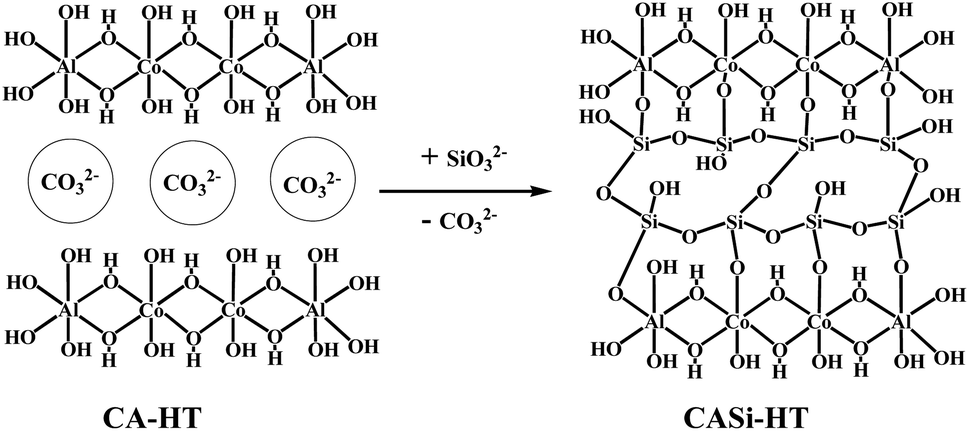 | ||
| Scheme 1 Schematic representation of intercalation on HT. | ||
Experimental section
Hydrotalcite (HT) with a Co–Al molar ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was synthesized according to a method previously described in the literature.24 Solution 1 was prepared by dissolving Co(NO3)2·6H2O (1 M) and Al(NO3)3·9H2O (0.33 M) in 2.6 L deionized H2O. Solution 2 was prepared by dissolving (NH4)2CO3 (0.67 M) and NH4OH (3.27 M) in 2.225 L deionized H2O. Solution 2 was added drop wise to solution 1 with constant stirring at 40 °C for 1 h, resulting in the precipitation of HT, with a final gel pH of 8.5. The gel was filtered and washed with deionized H2O until the pH of the filtrate reached 7.0. The sample was dried at 80 °C overnight and calcined at 400 °C in the presence of air for 6 h; this sample is represented as CA-Si0.
:1 was synthesized according to a method previously described in the literature.24 Solution 1 was prepared by dissolving Co(NO3)2·6H2O (1 M) and Al(NO3)3·9H2O (0.33 M) in 2.6 L deionized H2O. Solution 2 was prepared by dissolving (NH4)2CO3 (0.67 M) and NH4OH (3.27 M) in 2.225 L deionized H2O. Solution 2 was added drop wise to solution 1 with constant stirring at 40 °C for 1 h, resulting in the precipitation of HT, with a final gel pH of 8.5. The gel was filtered and washed with deionized H2O until the pH of the filtrate reached 7.0. The sample was dried at 80 °C overnight and calcined at 400 °C in the presence of air for 6 h; this sample is represented as CA-Si0.
Silicate anion intercalated HT was produced by introducing various concentrations of sodium silicate into pre-prepared HT. Silicate solutions of appropriate concentrations were added slowly to the HT gel at room temperature and stirred for 48 h. The silicate intercalated HT materials were then filtered and washed with deionized H2O. The intercalated HT samples prepared by using 1.42 and 2.03 M sodium silicate solutions are represented by CA-Si4 and CA-Si6, respectively and these samples calcined at 400 °C for 6 h in air. Further, attempts were made to retain the layered structure on silicate anion intercalated CoAl-HT by crystallizing the interlayer silicate anion species using a dry gel method. Approximately 1.5 g of as-synthesized CA-Si6 was wetted with the help of a calculated amount of 40% tetrapropylammonium hydroxide (TPAOH) solution. The resultant solid cake was allowed to crystallize at 175 °C for 72 h. The TPAOH was removed by calcination at 550 °C in air for 6 h. The interlayer silicate anion crystallized sample synthesized using CA-Si6 with TPAOH is represented as CA-Si6S. For the reference, CoAl2O4 prepared by solid state mixing of corresponding salts followed by calcination at 600 °C in air for 6 h.
Characterization
Powder XRD was performed to determine the bulk crystalline phases of the materials using a 18 KW XRD Rigaku, Japan with Cu-Kα radiation (λ = 1.54184 Å). The diffraction patterns were recorded in the 2θ range 5–70°, with a scan speed and step size of 0.5° min−1 and 0.02°, respectively. FT-IR spectra were obtained using a Perkin-Elmer 2000-FTIR in the range 400–4000 cm−1 using KBr pellets. The morphology and size of the materials were analysed using a Phillips Technai G2T30 TEM operated at 300 kV. The textural properties (BET surface area, micropore area (t-plot method), BJH average pore volume) of the samples were derived from N2 adsorption–desorption measurements carried out at −196 °C out using an automatic micropore physisorption analyser (Micromeritics ASAP 2020, USA) after the samples were degassed at 250 °C for at least 8 h under 10−3 Torr pressure prior to each run. Diffuse reflectance UV-Visible Spectra were recorded at 350–800 nm using Analytik jena, specord plus spectrometer with the help of BaSO4 as reference.X-ray photoelectron spectra (XPS) of the catalysts were recorded with custom built ambient pressure photoelectron spectrometer (APPES) (Prevac, Poland), equipped with VG Scienta's R3000HP analyzer and MX650 monochromator.27 Monochromatic Al Kα X-ray was generated at 450 W and used for measuring X-ray photoelectron spectrum (XPS) of the above samples. Base pressure in the analysis chamber was maintained in the range of 2 × 10−10 Torr. The energy resolution of the spectrometer was set at 0.7 eV at a pass energy of 50 eV. Binding energy (BE) was calibrated with respect to Au 4f7/2 core level at 84.0 eV. The error in the reported BE values is within 0.1 eV. The acidity of materials were studied as per the literature procedure28 by pyridine vapour adsorption, which was carried out in a Harrick Scientific HVC-DR2 reaction chamber with a detachable ZnSe window. About 100 mg (10% of sample was mixed with KBr) of sample was placed in the sample cup and was pre-activated at 350 °C for 6 h. Pyridine adsorption was done at 150 °C for 1 h under flowing helium. After attaining saturation desorption was done at different temperature. The amount of soluble basic sites was determined by acid–base titration method.29 In this method 100 mg of pure and silicate intercalated HT samples were vigorously shaken with 10 ml water about 24 h in room temperature and catalyst was separated. The filtrate was neutralized with 0.05 M of HCl. The remaining acid was titrated with 0.1 M of standard NaOH. Elemental composition present in the final materials were determined using Wavelength Dispersive X-ray Fluorescence spectrometer (WDXRF; ZSX PRIMUS Primus, Rigaku). Calibration of the equipment was carried out using standards containing Co, Al & Si in different proportions. Net intensity (peak–background) of standards was used for calibration and Co, Al & Si contents in the catalyst samples were obtained from the calibration curve.
Catalytic study
The catalytic activity of synthesized CoAl-HT and silicate anion intercalated HT were studied for the oxidation of various primary and secondary alcohols using t-butyl hydro peroxide (TBHP) as co-oxidant. For comparison, cobalt supported on silica and alumina was prepared by solid state mixing of cobaltous nitrate with silica and alumina support over the period of 3 h followed by calcination of 550 °C. The catalytic activity of Co/SiO2 and Co/Al2O3 was studied under identical conditions. The products were extracted with iso-propyl alcohol and analyzed by gas chromatography (Agilent Technologies 7890A, USA) using a flame ionization detector with an HP-5 column.Results and discussion
FT-IR spectra of as-synthesized hydrotalcite and silicate intercalated hydrotalcite are shown in Fig. 1. The FT-IR spectra are composed of absorption bands around 3450 cm−1 (not shown) and around 1600 cm−1 due to the water molecule present in the interlayer of hydrotalcite. CoAl-HT showed an absorption band of around 1350 cm−1 as a response to the stretching vibration interlayer CO32− anion.8,17 Silicate anion intercalated samples show a sharp new band appearing at around 1000 cm−1 due to the presence of Si–O–Si stretching and a band 800 cm−1 also due to Si–O stretching frequency.17,26 The observed broadening of the vibrational band near 1000 cm−1 along with shoulder peak around 1200 cm−1 indicates that the Si–O–Si bond angle is 180°, which is due to the presence of layered silicate anions in a distorted tetrahedral environment.17c An increase in the concentration of silicate anions in the gel resulted in a decrease in the intensity of the vibration band that corresponds to CO32− ions.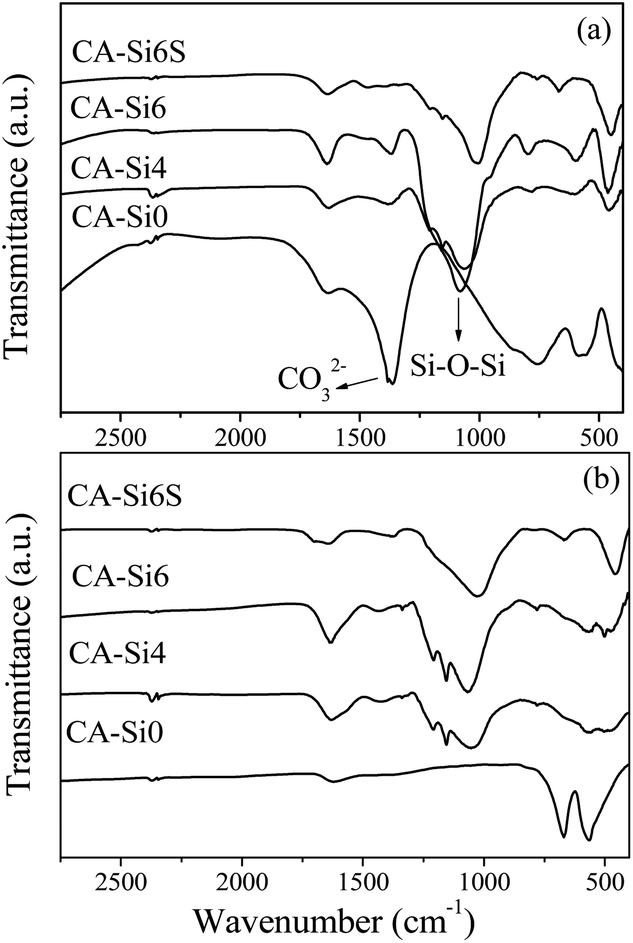 | ||
| Fig. 1 FT-IR spectra of as-synthesized (a) and calcined (b) HT and silicate intercalated materials. | ||
These results indicate that silicate anions were successfully intercalated between the brucite-like layers.26 After calcination, the FT-IR spectrum of CoAl-HT shows that a strong and sharp peak around 670 and 560 cm−1 indicates Co–O bonds of cobalt spinel (Co3O4).23,25,30 In silicate anion intercalated hydrotalcite, the relative intensity for the band 670 and 560 cm−1 was reduced and showed an intense peak around 1000 cm−1 corresponding to silicate anion, which supports the formation of layered cobalt hydrotalcite and cobalt silicate solid solution upon calcination. Further, the band at 470 cm−1 corresponding to Co–O–Si bending vibration indicates the presence of cobalt silicate species.31,32
Powder X-ray diffraction patterns of as-synthesized hydrotalcite and silicate intercalated hydrotalcite samples are shown in Fig. 2. CoAl-HT shows that sharp and symmetrical reflections of (003) and (006) planes around 2θ ranges 11 and 23, respectively, are typical of layered HT structure.7,25,26,30,31 The silicate anion intercalated hydrotalcite CA-Si4 and CA-Si6 showed broad peak evidence that the silicates are stuffed in the interlayer spaces of the brucite structure. Unlike earlier report, the current study it was not observed much shift in XRD d-spacing upon silicate intercalation. However, it is known that the peak position and broadening of X-ray reflection is directly reflects the orientation of anions present in the interlayer of HT.17 The broadening of X-ray reflection (Fig. 2b) evidently supports the orientation of the silicate anion in the interlayer as a layered silicate.17 Pure CoAl-HT (CA-Si0) shows an XRD pattern characteristic of cobalt spinel (Co3O4 and CoAl2O4) structure (Fig. 2b) upon calcination,25 whereas silicate anion intercalated CoAl-HT (CA-Si) resulted solid solution of cobalt silicates and cobalt spinel structure, which is in agreement with FT-IR studies. The stabilization of silicate anions by TPAOH results in a mesoporous solid solution of cobalt silicate and cobalt spinel structures.
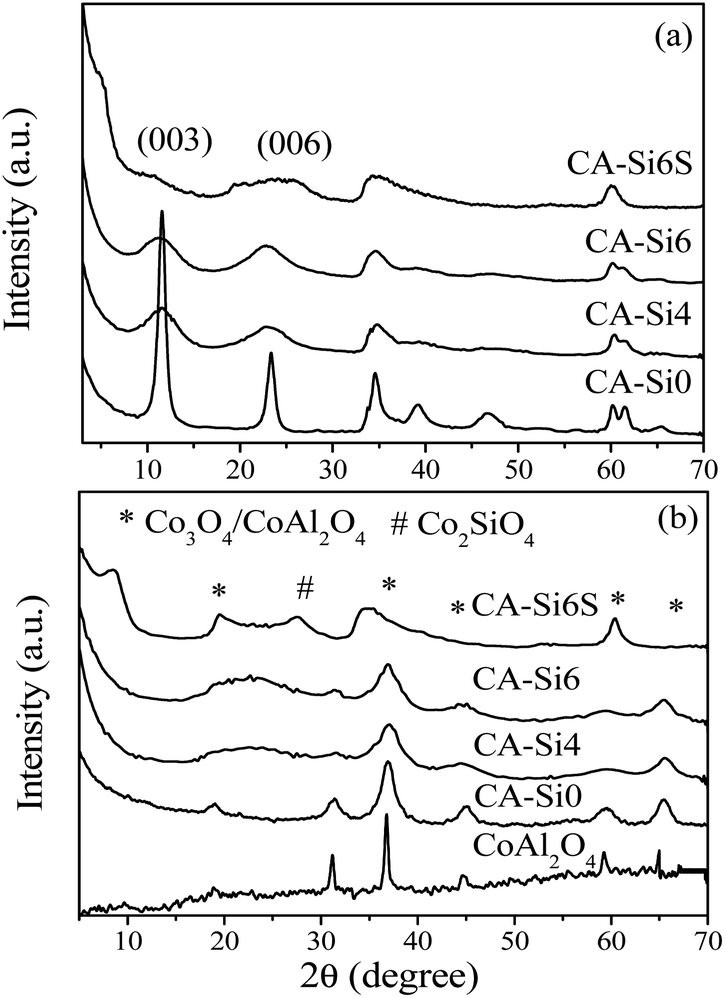 | ||
| Fig. 2 XRD patterns of (a) as-synthesized and (b) calcined silicate free HT and silicate intercalated materials. | ||
The elemental composition of CoAl-HT and silicate intercalated HT were estimated using WDXRF and results are shown in Table S1 (ESI†). It was evident that slight decrease in cobalt content in final materials is noticed as compared to synthesis gel composition. This might be due to an incomplete condensation of cobalt species, which remain as soluble cobalt species during the synthesis washed while filtering.
Nitrogen sorption isotherms of CoAl-HT and silicate intercalated HT correspond to multilayer adsorption followed by capillary condensation typically of type IV isotherms (Fig. 3) according to the IUPAC.33,34 Textural properties of various cobalt hydrotalcite samples are summarized in Table 1. The BET surface area and pore volume of the silicate-intercalated HT samples were found to be higher than those of the pure HT sample. The observed increase in surface area in CA-Si samples are owing to intercalation of silicate anion by replacing carbonate anions is in agreement with the FT-IR spectra (Fig. 1).
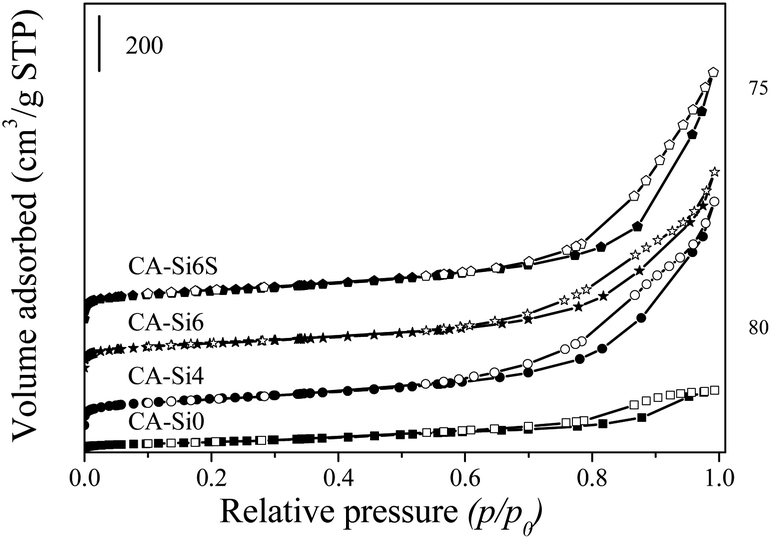 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms of various hydrotalcite samples. | ||
| Sample code | Calcination temp.(°C) | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Basicity (mmol g−1) | ||
|---|---|---|---|---|---|---|
| BET | Micropore (t-plot) | Total | Micropore | |||
| CA-Si0 | 400 | 142 | 0 | 0.35 | 0 | 2.85 |
| CA-Si4 | 400 | 342 | 29.0 | 1.20 | 0.012 | 2.94 |
| CA-Si6 | 400 | 317 | 33.5 | 1.11 | 0.015 | 4.0 |
| CA-Si6S | 550 | 361 | 54.5 | 1.40 | 0.026 | — |
A further increase in the anion concentration (2.03 M; CA-Si6) resulted in a slight decrease in the surface area (317 m2 g−1) and the pore volume. These reductions were a result of the filling of the HT interlayers by condensed polymeric silicates in distorted environment.17 Treatment with TPAOH followed by calcination at 550 °C resulted in an increase in porous nature of silicate anion present on interlayer, which enhances surface area and micropore volume, support the formation of porous layered pillars between the HT layers, as the results are in agreement with powder XRD. The surface acidity of HT and silicate intercalated HT were followed by pyridine-FTIR spectroscopy and the results are shown in Fig. S1 (ESI†). The pure cobalt HT show absorption bands at 1449 cm−1 which can be attributed to pyridine coordinated to Al3+ ions.35 As the desorption temperature increases from 200 to 500 °C, the vibrational band corresponding to Lewis acid sites remains accountable indicate that presence of Al3+ in the surface and has strong interaction with pyridine. In contrast, in the silicate anion intercalated HT (CA-Si6) the peak at 1449 cm−1 decreases drastically with desorption temperature evident the presence of weak Lewis acidity on the surface. Further, an additional broad absorption band around 1596 cm−1 was evident due to presence of surface hydroxyl group attached to cobalt and silica species.35
The amount of soluble basic sites was measured by acid–base titration method,29 after the samples were treated with 0.05 M HCl and results are shown in Table 1. It was evident from the above results, the amount of surface basic sites increases with increase in silicate anion concentration. The increase in surface basicity on silicate anion intercalated sample is due to exposed more oxide species (Co2+ and Co3+) present on the surface compared to silicate anion free HT.36 The TEM images of pure HT and silicate intercalated HT samples are shown in Fig. 4. In pure HT (CA-Si0) sample shows non-uniform elongated spherical shape morphology with clear lattice fringes typical of spinel structure25e (further supported from ED pattern Fig. S2a†), which is in agreement with XRD. The introduction of silicate anion into the interlayer of HT resulted to formation of crumbled hexagonal plates. The ED pattern of calcined CoAl-HT-Si (Fig. S2b†) showed characteristic lattice feature of hexagonal cobalt silicate31 and spinel structure.25c It supports the intercalation of silicate anion in the interlayer and formed the solid solution of cobalt silicate and cobalt spinel as evident in XRD. The coordination and oxidation status of cobalt species present in the structure were followed with the help of DR UV-Vis spectra (Fig. 5). All the as-synthesised samples show (Fig. 5a) broad band near to 530 nm related to 4T1g (F) → 4T1g (P) corresponds to Co2+ in octahedral (Oh) environment.30b The absorption band in the range of around 350–400 nm region are assigned to typical of Co3+ (HT; 1A1g → 1T1g).32 Further, the presence of band around 430 nm could be assigned to d–d transitions (1A1g → 1T2g) of Co3+ (Oh) species incorporated in the octahedral sites of Al2O3 matrix.30b,32 After calcination (see Fig. 5b) the broadening of band around 680 nm on typical of Co2+ in tetrahedral environment in CoAl2O4.32 In addition, the silicate anion stabilized HT (CA-Si6S) exhibit broad triplet absorption bands (540, 580, and 626 nm) are assigned to the 4A2(F) → 4T1(P) transition of Co(II) in the tetrahedral silicate environment,32c further support the formation solid solution of cobalt silicate and spinel structure (Fig. 5).
 | ||
| Fig. 4 TEM images of CA-Si0 (a), CA-Si4 (b), CA-Si6 (c) and CA-Si6S (d). | ||
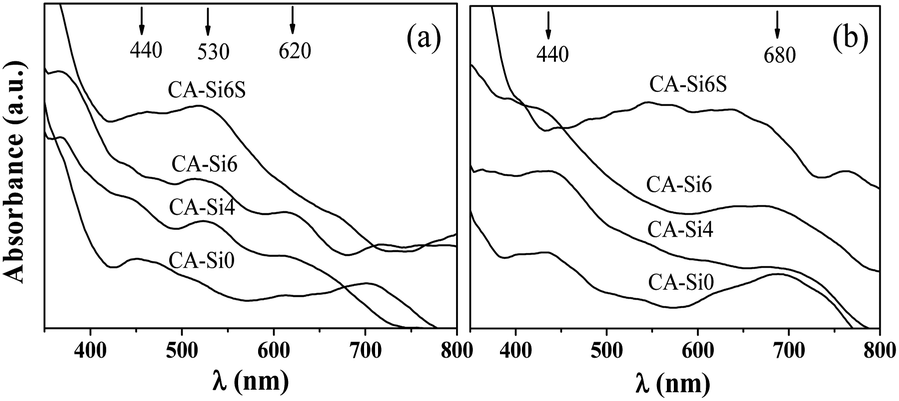 | ||
| Fig. 5 DR UV-Vis spectra of as-synthesized (a) and calcined (b) HT and silicate intercalated materials. | ||
In order to understand the nature of the active species present on the surface, XPS has been carried out for silicate free cobalt hydrotalcite (CA-Si0) and silicate intercalated hydrotalcite (CA-Si4, CA-Si6) and spent catalyst (CA-Si6R). The important features of the XPS data are summarized in Table 2 and Co 2p core level spectra of these samples are shown in Fig. 6. XPS of Co 2p shows two components appearing due to spin–orbital splitting of Co 2p3/2 and Co 2p1/2 peaks with binding energies around 782 and 797 eV along with satellites indicating cobalt is predominantly in mixed oxidation state on the surface.25b The binding energy difference between Co 2p1/2 and Co 2p3/2 is about 15.2 ± 0.1 eV for pure hydrotalcite further support the presence of Co3+ on the surface.25b Due to strong interaction of Co3+ on the surface in the form of CoO(OH) species (Fig. 6).25b
| Catalysts | Co 2p3/2 (eV) | Co 2p1/2 (eV) | Spin orbital splitting (eV) | Difference between main and satellite peaks for | O1s (eV) | Al 2p (eV) | Si 2p (eV) | Si/Co ratio | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Main | Satellite | Main | Satellite | Co 2p3/2 (eV) | Co 2p1/2 (eV) | Surface XPS | Bulk XRF | |||||
| CA-Si0 | 782.2 | 788.5 | 797.3 | 804.9 | 15.1 | 6.3 | 7.6 | 533.0 | 75.9 | — | — | — |
| CA-Si4 | 781.8 | 787.8 | 797.1 | 804.2 | 15.3 | 6.0 | 7.1 | 532.5 | 74.6 | 103.5 | 0.18 | 0.51 |
| CA-Si6 | 781.3 | 788.5 | 796.9 | 804.3 | 15.6 | 7.2 | 7.4 | 532.5 | 74.3 | 103.4 | 0.36 | 0.71 |
| CA-Si6R | 781.5 | 787.2 | 797.0 | 803.7 | 15.5 | 5.7 | 6.7 | 532.7 | 74.6 | 103.5 | — | — |
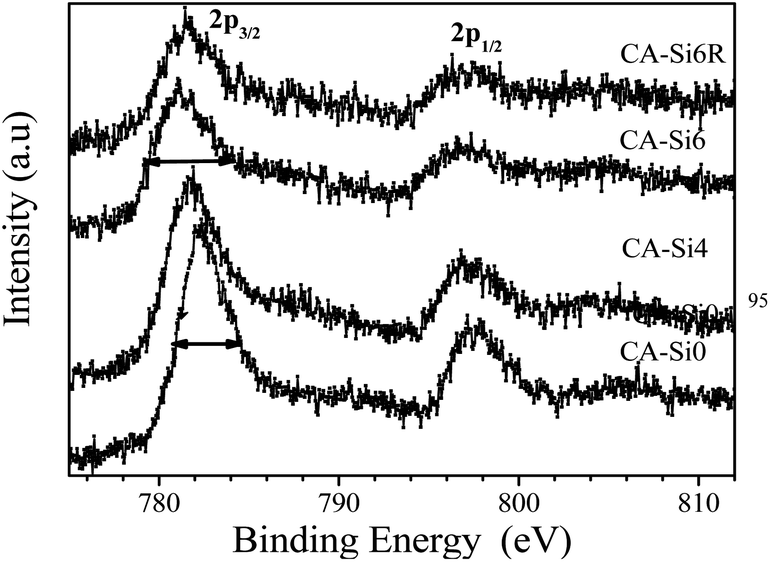 | ||
| Fig. 6 XPS of Co 2p in various HT materials. | ||
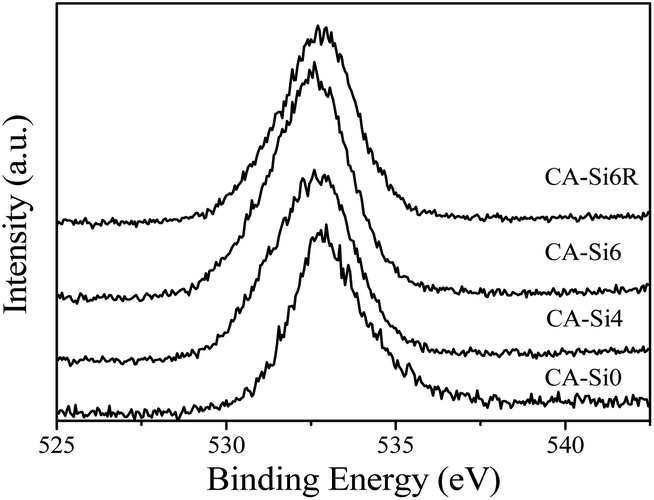 | ||
| Fig. 7 XPS of O 1s in various HT materials. | ||
On silicate intercalation, there is a broadening observed, especially, on the lower binding energy side and it is indicated by solid arrows on CA-Si0 and CA-Si6. Above broadening is accompanied with an increase in spin-orbit splitting from 15.3 to 15.5 eV in the silicate anion intercalated species and regenerated catalysts indicates the presence of Co2+ ions with possible formation of cobalt aluminates and silicates species.38 This is also supported by the satellite lines which are located at about 6–7 eV above the photoline corresponds to presence of high-spin Co2+ compounds such as CoO, CoAl2O4 and CoSi2O5 species.39 The XPS core level spectra of Co 2p3/2 were further deconvoluted using XPS Peak 4.1 software with Shirley background and using Gaussian–Lorentzian fitting to quantify the various oxidation state present in the materials and the results are shown in Table 3. The deconvoluted XPS CA-Si0 spectrum showed a major peak at 781.9 and less intense peak at 780.7 are typical of cobalt in Co2+ and Co3+ oxidation state with cobalt aluminate (CoAl2O4) species.40 In the case of silicate intercalated samples peaks are broader (indicated by solid line); the deconvolution shows two major peaks at 781.9 eV and 780.7 eV which are typical of Co2+ in oxides in the form of Cobalt oxides, cobalt silicates, cobalt aluminates and cobalt in a cobalt oxides, where cobalt has an oxidation state of Co3+.40 The relative ratio Co3+/Co2+ species increases on silicate intercalated samples to about 0.55–0.65 indicating the silicate intercalated (CoAl-HTSi) samples exposes mixed oxidation states of Co by nearly equivalent amount.
The surface composition obtained from the XPS studies showed Si/Co ratio of 0.36 and 0.18 respectively. However the bulk composition of Si/Co on silicate intercalated samples calculated using XRF found to be 0.71 and 0.51 respectively for CA-Si6 and CA-Si4 samples. The presence of less silica concentration on the surface supports that most of the silicate anions are intercalated between the layers of HT and formed solid solution.
The O 1s spectrum of CA-Si0 (see Fig. 7) exhibit a peak around 533 eV and the corresponding silicate anion intercalated cobalt aluminium HT (CA-Si4, CA-Si6 and CA-Si6R) exhibits (Fig. 7) lower binding energy values around 532.5 eV. As in Co 2p core level, there is a clear broadening observed on the lower binding energy side and it is attributed to the presence of more than one kind of oxygen present on the surface. Especially, oxygen from silicate and hydroxyl are known to be observed between 532 and 533 eV. The Al 2p and Si 2p core level peak (figure shown in S3a and S3b (ESI†)) for silicate anion intercalated HT showed at 74 and 103–104 eV, respectively, which is significantly higher than that of pure Al2O3 and SiO2 support, indicate the presence of strong interaction between cobalt with alumina and silicate species. Further the observed shift in Al 2p XPS value on silicate intercalated HT revealed that Al present in distorted Td environment.37
The catalytic activities of CoAl-HT (CA-Si0), CoAl-HTSi (CA-Si4, CA-Si6), and silicate-anion-stabilized CoAl-HTSi (CA-Si6S) in the oxidation of benzhydrol to benzophenone at 70 °C for 6 h were investigated; the results are summarized in Table 4. Although CoAl-HT intercalated with silicate ions had lower cobalt content than pure CoAl-HT, it gave a higher yield of benzophenone. The higher yield obtained on CA-Si6 was the result of the presence of active cobalt species (Co2+ and Co3+) exposed on the surface of the hydrotalcite layer. These results are in agreement with the basicity and XPS results. The high turn over number (TON) on CA-Si6 compared with that on silicate-free CoAl-HT further supports the presence of exposed cobalt species on the surface. In all cases, benzophenone was obtained as the major product, along with a small amount of a peroxo intermediate. To understand the mechanistic pathway, the reaction was carried out in the presence of a radical scavenger, i.e. 2, 6-di-tert-butyl-p-cresol (BHT). It was found that in the presence of BHT, the conversion decreased to 45%. It is therefore clear that reaction proceeds through a free-radical mechanism. The proposed mechanism, based on surface-active species identified in XPS studies, is shown in Scheme 2.41 First, tert-butyl hydro peroxide (TBHP) interacts with surface-active Co3+ and Co2+ species, yielding peroxo and alkoxo radicals. The peroxo radical interacts with the alcohol, with abstraction of the proton present at the benzylic position, and forms the corresponding radical. The resultant radical interacts with the already-formed alkoxo radical, resulting in production of a ketone or aldehyde, along with tert-butyl alcohol as a by-product. The CA-Si6 catalyst was re-used after separation from the reaction mixture; it was found that the catalytic activity was intact. However, the silicate-intercalated CA-Si6 was regenerated by recalcination at 550 °C in order to remove any chemisorbed organic compounds, and the reaction over the regenerated catalyst was studied. It was again found that the catalytic conversion remain constant with decrease in benzophenone selectivity.
| S. No | Catalyst | Co contentb (Wt%) | Conv. (%) | Sel. (%) | Yield (%) | TONc |
|---|---|---|---|---|---|---|
| a Reaction conditions: alcohol:oxidant mole ratio of 1:1; temperature = 70 °C; time = 6 h.b Analysed using Atomic Absorption Spectrometer (AAS).c Turn over number (TON) calculated based on molar content of Co present in the catalyst.d Recycled catalyst without calcination.e Recycled catalyst 550 °C calcined.f 2 mmole of BHT radical scavenger used. |
||||||
| 1 | Without catalyst | 0.0 | 18.0 | 100 | 18.0 | — |
| 2 | CA-Si0 | 38 | 73.2 | 100 | 73.2 | 1144 |
| 3 | CA-Si4 | 24 | 83.8 | 100 | 83.8 | 2095 |
| 4 | CA-Si6 | 18 | 85.7 | 99.6 | 85.3 | 2856 |
| 5 | CA-Si6d | 18 | 81.5 | 100 | 81.5 | — |
| 6 | CA-Si6e | 18 | 87.5 | 70.0 | 61.2 | 2766 |
| 7 | CA-Si6f | 18 | 45.2 | 100 | 45.2 | — |
| 8 | CA-Si6S | — | 82.7 | 96.8 | 80.0 | — |
| 9 | Co/Al2O3 | 18 | 23.0 | 100 | 23.0 | — |
| 10 | Co/SiO2 | 18 | 11.7 | 100 | 11.7 | — |
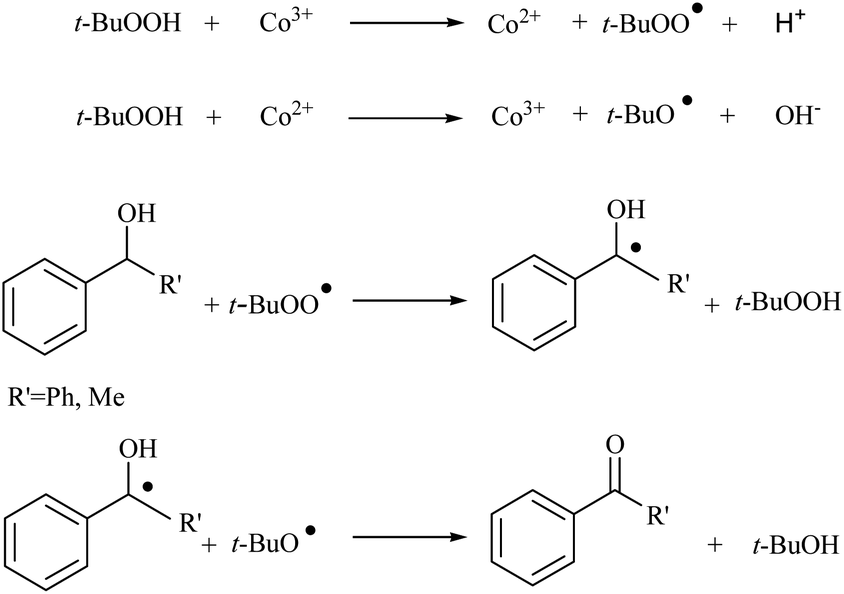 | ||
| Scheme 2 Plausible mechanism for oxidation of alcohol. | ||
The XPS core cobalt 2p level spectrum of the regenerated catalyst was found to be very similar to that of the fresh catalyst, showing that the catalytic activity was stable. The catalytic activity was also compared with those of samples prepared by physical mixing, i.e. Co/SiO2 and Co/Al2O3. The activities of the physical mixtures were much lower than that of the silicate-anion-intercalated CA-Si6 sample; this is because active cobalt species are uniformly distributed on the catalyst surface (Table 4). The highly active CA-Si6 catalyst was further studied in oxidation of a series of substituted aromatic and aliphatic alcohols; the results are shown in Table 5. The catalyst gave very good yields in all cases. The catalyst also gave very good conversion in the oxidation of aliphatic alcohols, clearly supporting the presence of large numbers of exposed active sites. However, alcohol conversion was lower in the presence of electron-withdrawing groups such as NO2− and Br− at the para position; this might be the result of stabilization of free radicals formed on the surface during the reaction. In contrast, the conversion decreased slightly in the presence of an electron-withdrawing group at the meta position and a methoxy group at the ortho position. Overall, the catalyst was found to be promising for several alcohol oxidations under ambient conditions.
| S. No | Substrate | Catalyst | Product | Conv. (%) | Sel. (%) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 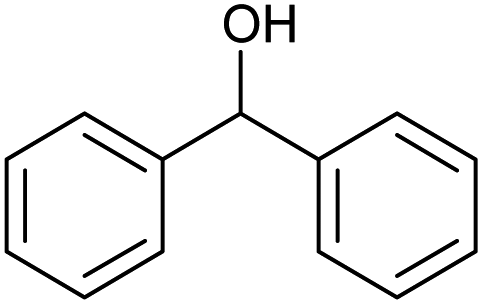 |
Without catalyst | 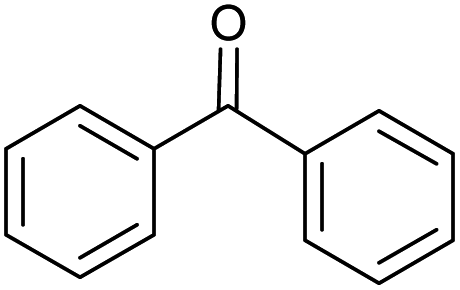 |
18.0 | 100 | 18 |
| 2 | 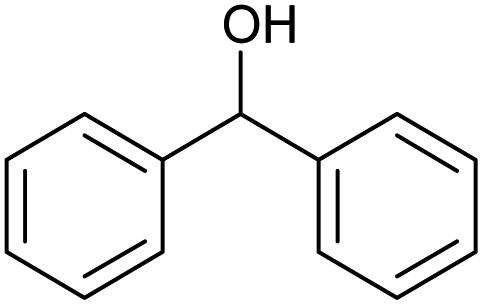 |
CA-Si6 | 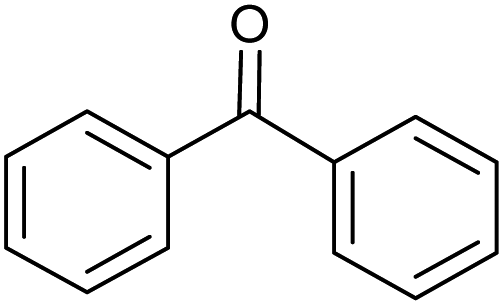 |
85.7 | 99.6 | 85.3 |
| 3 | 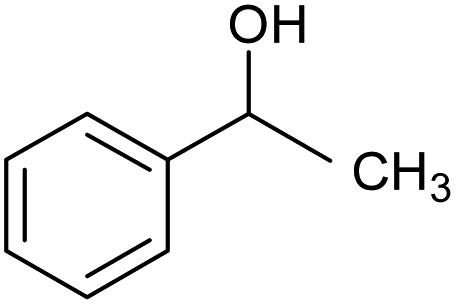 |
CA-Si6 | 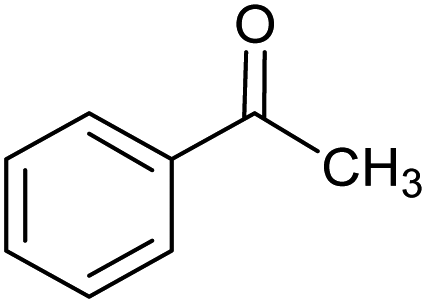 |
80.1 | 95.7 | 76.6 |
| 4 | 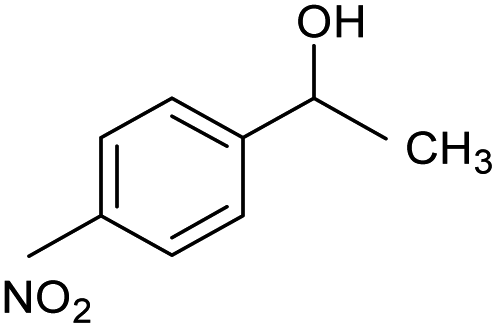 |
CA-Si6 | 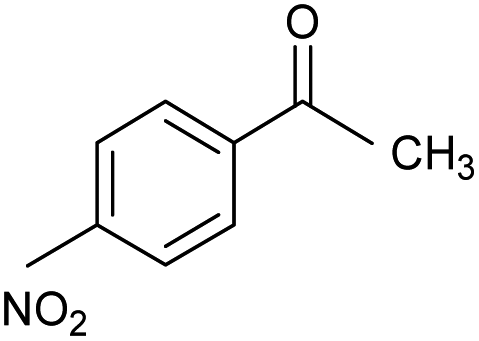 |
66.5 | 96.5 | 64.2 |
| 5 | 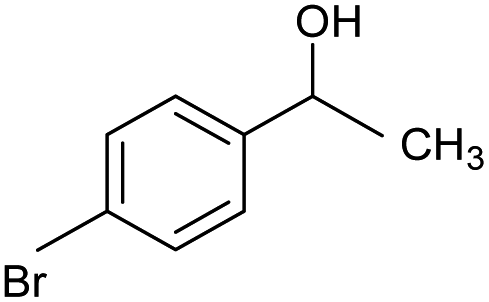 |
CA-Si6 | 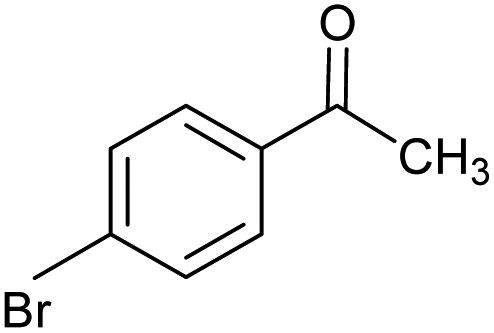 |
68.0 | 96.0 | 65.3 |
| 6 | 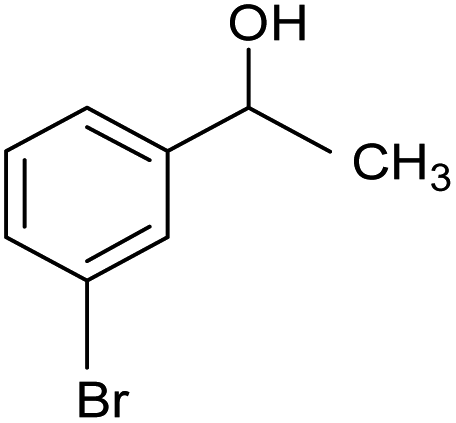 |
CA-Si6 | 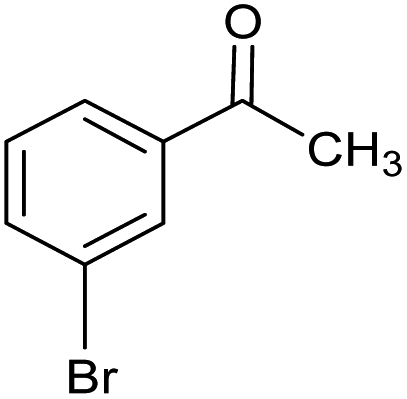 |
74.7 | 77.6 | 58.0 |
| 7 |  |
CA-Si6 |  |
78.2 | 77.6 | 60.7 |
| 8 |  |
CA-Si6 |  |
90.3 | 97.0 | 87.5 |
Conclusions
In summary, nano-sized silicate anion intercalated CoAl-HT was synthesized. FT-IR, XRD, N2 sorption and DR-UV-Vis studies revealed that CoAl-HTSi formed a solid solution of cobalt spinel and cobalt silicate. The presence of cobalt in divalent and trivalent oxidation state was evident from XPS studies. The silicate intercalated sample shows relatively more divalent cobalt species and have strong interaction with Si and Al species. The resultant materials are shown as promising catalysts for oxidation of various alcohols.Acknowledgements
AS extend his sincere and grateful thanks to the University of Delhi and CSIR (project no: 01(2516)/11/EMR-II), New Delhi for the financial supports. Authors also express sincere thanks to USIC, Delhi University, Heterogeneous catalysis and reaction engineering laboratory, IIT-Delhi for their support on instrumentation facility. Authors acknowledge the help to record XPS data at Center of Excellence on Surface Science at CSIR-NCL, Pune, and Dr C. S. Gopinath for his valuable suggestions and discussion.References
- (a) S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17 RSC; (b) M. L. Kantam, U. Pal, B. Sreedhar, S. Bhargava, Y. Iwasawa, M. Tada and B. M. Choudary, Adv. Synth. Catal., 2008, 350, 1225 CrossRef CAS; (c) G. Tojo and M. Fernandez, in Oxidation of alcohols to aldehydes and ketones: a guide to current common practice, Springer, New York, NY, USA, 2006 Search PubMed; (d) D. Lenori, Angew. Chem., Int. Ed., 2006, 45, 3206 CrossRef PubMed; (e) T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS PubMed; (f) B. Z. Zhan and A. Thompson, Tetrahedron, 2004, 60, 2917 CrossRef CAS PubMed.
- K. Layek, H. Maheswaran, R. Arundhathi, M. L. Kantam and S. K. Bhargava, Adv. Synth. Catal., 2011, 353, 606 CrossRef CAS.
- Y. M. A. Yamada, T. Arakawa, H. Hocke and Y. Uozumi, Chem. - Asian J., 2009, 4, 1092 CrossRef CAS PubMed.
- (a) Y. M. A. Yamada, T. Arakawa, H. Hocke and Y. Uozumi, Angew. Chem., Int. Ed., 2007, 46, 704 CrossRef CAS PubMed; (b) T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Adv. Synth. Catal., 2009, 351, 1890 CrossRef CAS; (c) M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127 CrossRef CAS; (d) P. Maity, C. S. Gopinath, S. Bhaduri and G. K. Lahiri, Green Chem., 2009, 11, 554 RSC.
- (a) P. Liu, Y. Guan, R. A. Van Santen, C. Li and E. J. M. Hensen, Chem. Commun., 2011, 47, 11540 RSC; (b) A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC; (c) M. L. Kantam, J. Yadav, S. Laha, B. Sreedhar and S. Bhargava, Adv. Synth. Catal., 2008, 350, 2575 CrossRef CAS.
- (a) M. B. Gawande, A. Rathi, I. D. Nogueira, C. A. A. Ghumman, N. Bundaleski, O. M. N. D. Teodoro and P. S. Branco, ChemPlusChem, 2012, 77, 865 CrossRef CAS; (b) C. I. Fernandes, C. D. Nunes and P. D. Vaz, Curr. Org. Synth., 2012, 9, 670 CrossRef CAS.
- (a) F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS; (b) F. Basile, G. Fornasari, M. Gazzano and A. Vaccari, J. Mater. Chem., 2002, 12, 3296 RSC; (c) P. Arpentinier, F. Basile, P. Del Gallo, G. Fornasari, D. Gary, V. Rosetti and A. Vaccari, Catal. Today, 2005, 99, 99 CrossRef CAS PubMed.
- (a) T. Lopez, P. Bosch, M. Asomoza, R. Gomez and E. Ramos, Mater. Lett., 1997, 31, 311 CrossRef CAS; (b) V. Davila, E. Lima, S. Bulbulian and P. Bosch, Microporous Mesoporous Mater., 2008, 107, 240 CrossRef CAS PubMed.
- (a) F. Malherbe and J. P. Besse, J. Solid State Chem., 2000, 155, 332 CrossRef CAS; (b) A. I. Khan and D. O'Hare, J. Mater. Chem., 2002, 12, 3191 RSC; (c) M. Adachi-Pagano, C. Forano and J. P. Besse, J. Mater. Chem., 2003, 13, 1988 RSC.
- (a) R. Ma and T. Sasaki, Recent Pat. Nanotechnol., 2012, 6, 159 CrossRef CAS; (b) Y. Lin and G. Wang, Recent Pat. Nanotechnol., 2012, 6, 169 CrossRef CAS.
- (a) Layered Double Hydroxides-Structure and bonding, ed. X. Duan and D. G. Evans, Springer-Verlag, Berlin, Germany, 2005, vol. 119 Search PubMed; (b) Z. Sun, L. Jin, S. He, Y. Zhao, M. Wei, D. G. Evans and X. Duan, Green Chem., 2012, 14, 1909 RSC.
- (a) K. Putyera, J. Jagiello, T. J. Bandosz and J. A. Schwarz, J. Chem. Soc., Faraday Trans., 1996, 92, 1243 RSC; (b) O. Meyer, F. Roessner, R. A. Rakoczy and R. W. Fischer, ChemCatChem, 2010, 2, 314 CrossRef CAS.
- (a) C. Misra and A. J. Perrotta, Clays Clay Miner., 1992, 40, 145 CAS; (b) K. Parida, M. Satpathy and L. Mohapatra, J. Mater. Chem., 2012, 22, 7350 RSC.
- (a) K. Takehira, T. Kawabata, T. Shishido, K. Murakami, T. Ohi, D. Shoro, M. Honda and K. Takaki, J. Catal., 2005, 231, 92 CrossRef CAS PubMed; (b) K. Takehira and T. Shishido, Catal. Surveys Asia, 2007, 11, 1 CrossRef CAS; (c) G. Centi and S. Perathoner, Microporous Mesoporous Mater., 2008, 107, 3 CrossRef CAS PubMed.
- (a) M. J. Climent, A. Corma, S. I. Borra, K. Epping and A. Velty, J. Catal., 2004, 225, 316 CrossRef CAS PubMed; (b) M. Bolognini, F. Cavani, D. Scagliarini, C. Flego, C. Perego and M. Saba, Microporous Mesoporous Mater., 2003, 66, 77 CrossRef CAS PubMed.
- (a) C. A. Antonyraj, P. Koilraj and S. Kannan, Chem. Commun., 2010, 46, 1902 RSC; (b) S. Velu, K. Suzuki, M. Okazaki, T. Osaki, S. Tomura and F. Ohashi, Chem. Mater., 1999, 11, 2163 CrossRef CAS; (c) B. F. Sels, D. E. De Vos and P. A. Jacobs, Catal. Rev.: Sci. Eng., 2001, 43, 443 CrossRef CAS PubMed.
- (a) A. Schutz and P. Biloen, J. Solid State Chem., 1987, 68, 360 CrossRef CAS; (b) S. K. Yun, V. R. L. Constantino and T. J. Pinnavaia, Clays Clay Miner., 1995, 43, 503 CAS; (c) C. Depege, F.-Z. EI Metoui, C. Forano, A. de Roy, J. Dupuis and J. P. Besse, Chem. Mater., 1996, 8, 952 CrossRef CAS; (d) M. A. Drezdon, US Pat.,4,774,212, 1988; (e) M. Foulon and L. Stelandre, US Pat.,7,985,390 B2, 2011.
- (a) M. del Arco, S. Gutierrez, V. Rives, R. Trujillano and P. Malet, Inorg. Chem., 1996, 35, 6362 CrossRef CAS PubMed; (b) V. Rives and M. A. Ulibarri, Coord. Chem. Rev., 1999, 181, 61 CrossRef CAS; (c) M. del Arco, S. Gutierrez, C. Martin, V. Rives and J. Rocha, J. Solid State Chem., 2000, 151, 272 CrossRef CAS; (d) D. Carriazo, C. Martin and V. Rives, Eur. J. Inorg. Chem., 2006, 22, 4608 CrossRef.
- (a) I. Carpani, M. Berrettoni, M. Giorgetti and D. Tonelli, J. Phys. Chem. B, 2006, 110, 7265 CrossRef CAS PubMed; (b) J. Twu and P. K. Dutta, J. Catal., 1990, 124, 503 CrossRef CAS; J. Twu and P. K. Dutta, Chem. Mater., 1992, 4, 398 Search PubMed; (c) U. Costantino, F. Montanari, M. Nocchetti, F. Canepa and A. Frache, J. Mater. Chem., 2007, 17, 1079 RSC; (d) S. Fleutot, J. C. Dubin, I. Baraille, C. Forano, G. Renaudin, F. Leroux, D. Gonbeau and H. Martinez, Phys. Chem. Chem. Phys., 2009, 11, 3554 RSC.
- (a) A. Kumar, K. Iwatani, S. Nishimura, S. Takagaki and K. Ebitani, Catal. Today, 2012, 185, 241 CrossRef CAS PubMed; (b) A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2011, 50, 2986 CrossRef CAS PubMed.
- (a) M. L. Kantam, A. Ravindra, Ch. V. Reddy, B. Sreedhar and B. M. Choudary, Adv. Synth. Catal., 2006, 348, 569 CrossRef CAS; (b) M. G. Álvarez, M. Plíšková, A. M. Segarra, F. Medina and F. Figueras, Appl. Catal., B, 2012, 113–114, 212 CrossRef PubMed.
- S. Abelló, S. Mitchell, M. Santiago, G. Stoica and J. Pérez-Ramírez, J. Mater. Chem., 2010, 20, 5878 RSC.
- (a) J. T. Kloprogge and R. I. Frost, J. Solid State Chem., 1999, 146, 506 CrossRef CAS; (b) S. J. Palmer and R. L. Frost, Ind. Eng. Chem. Res., 2010, 49, 8969 CrossRef CAS.
- J. Shen, J. M. Kobe, Y. Chen and J. A. Dumesic, Langmuir, 1994, 10, 3902 CrossRef CAS.
- (a) J. Perez-Ramirez, A. Ribera, F. Kapteijn, E. Coronado and C. J. Gomez-Garcia, J. Mater. Chem., 2002, 12, 2370 RSC; (b) S. Kannan and C. S. Swamy, Catal. Today, 1999, 53, 725 CrossRef CAS; (c) D. Barreca, A. Gasparotto, O. I. Lebedev, C. Maccato, A. Pozza, E. Tondello, S. Turner and G. V. Tendeloo, CrystEngComm, 2010, 12, 2185 RSC; (d) T. Mathew, M. Vijayaraj, S. Pai, B. B. Tope, S. G. Hegde, B. S. Rao and C. S. Gopinath, J. Catal., 2004, 227, 175 CrossRef CAS PubMed; (e) L. Bruce, J. Takos and T. W. Turney in Novel Materials in Heterogeneous Catalysis, ed. R. T. K. Baker and L. L. Murrell, ACS Symposium Series, 1990, vol. 437, ch.13, pp. 129–139 Search PubMed.
- T. Baskaran, R. Kumaravel, J. Christopher and A. Sakthivel, RSC Adv., 2013, 3, 16392 RSC.
- (a) K. Roy, C. P. Vinod and C. S. Gopinath, J. Phys. Chem. C, 2013, 117, 4717 CrossRef CAS; (b) S. Velu, K. Suzuki, C. S. Gopinath, H. Yoshida and T. Hattori, Phys. Chem. Chem. Phys., 2002, 4, 1990 RSC.
- (a) A. K. Singh, R. Yadav and A. Sakthivel, Microporous Mesoporous Mater., 2013, 181, 166 CrossRef CAS PubMed; (b) R. Yadav, A. K. Singh and A. Sakthivel, Chem. Lett., 2013, 42, 1160 CrossRef CAS.
- R. Devi, R. Borah and R. C. Deka, Appl. Catal., A, 2012, 433–434, 122 CrossRef CAS PubMed.
- (a) X. Xie, K. Yan, J. Li and Z. Wang, Catal. Commun., 2008, 9, 1128 CrossRef CAS PubMed; (b) M. Gabrovska, R. Edreva-Kardjieva, K. Tenchev, P. Tzvetkov, A. Spojakina and L. Petrov, Appl. Catal., A, 2011, 399, 242 CrossRef CAS PubMed.
- H. Cui, M. Zayat and D. Levy, J. Sol-Gel Sci. Technol., 2006, 40, 83 CrossRef CAS.
- (a) M. A. Ulibarri, J. M. Fernandez, F. M. Labajos and V. Rives, Chem. Mater., 1991, 3, 626 CrossRef CAS; (b) J. P. Ramírez, G. Mul, F. Kapteijn and J. A. Moulijn, Mater. Res. Bull., 2001, 36, 1767 CrossRef; (c) P. Selvam and S. K. Mohapatra, J. Catal., 2005, 233, 276 CrossRef CAS PubMed; (d) L. G. A. van de Water, G. L. Bezemer, J. A. Bergwerff, M. V. Helder, B. M. Weckhuysen and K. P. de Jong, J. Catal., 2006, 242, 287 CrossRef CAS PubMed.
- M. Kruck and M. Jaroniec, Chem. Mater., 2001, 13, 3169 CrossRef.
- M. L. Occelli, J. P. Olivier, A. Auroux, M. Kalwei and H. Eckert, Chem. Mater., 2003, 15, 4231 CrossRef CAS.
- (a) G. A. H. Mekhemer, H. M. M. Abd-Allah and S. A. A. Mansour, Colloids Surf., A, 1999, 160, 251 CrossRef CAS; (b) T. Mathew, B. B. Tope, N. R. Shiju, S. G. Hegde, B. S. Rao and C. S. Gopinath, Phys. Chem. Chem. Phys., 2002, 4, 4260 CAS.
- N. Bahlawane, P. H. T. Nagmou, V. Vannier, T. Kottke, J. Heberle and K. K. Höinghaus, Phys. Chem. Chem. Phys., 2009, 11, 9224 RSC.
- (a) S. Velu, K. Suzuki and C. S. Gopinath, J. Phys. Chem. B, 2002, 106, 12737 CrossRef CAS; (b) T. Mathew, B. S. Rao and C. S. Gopinath, J. Catal., 2004, 222, 107 CrossRef CAS PubMed; (c) T. Mathew, K. Sivaranjani, E. S. Gnanakumar, Y. Yamada, T. Kobayashi and S. Gopinath, J. Mater. Chem., 2012, 22, 13484 RSC; (d) V. L. Parola, G. Deganello, S. Scirè and A. M Venezia, J. Solid State Chem., 2003, 174, 482 CrossRef.
- (a) S. B. Waghmode, R. Vetrivel, S. G. Hegde, C. S. Gopinath and S. Sivasanker, J. Phys. Chem. B, 2003, 107, 8517 CrossRef CAS; (b) S. G. Gholap, M. V. Badiger and C. S. Gopinath, J. Phys. Chem. B, 2005, 109, 13941 CrossRef CAS PubMed; (c) K. Sivaranjani, A. Verma and C. S. Gopinath, Green Chem., 2012, 14, 461 RSC.
- A. A. Khassin, T. M. Yurieva, V. V. Kaichev, V. I. Bukhtiyarov, A. A. Budneva, E. A. Paukshtis and V. N. Parmon, J. Mol. Catal. A: Chem., 2001, 175, 189 CrossRef CAS.
- (a) V. M. Jiménez, J. P. Espinós and A. R. González-Elipe, Surf. Interface Anal., 1998, 26, 62 CrossRef; (b) Q. Tang, Q. Zhang, P. Wang, Y. Wang and H. Wan, Chem. Mater., 2004, 16, 1967 CrossRef CAS.
- J. Taghavimoghaddam, G. P. Knowles and A. L. Chaffee, Top. Catal., 2012, 55, 571 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 pyridine FT-IR spectra of CA-HT and CA-Si6 at different desorption temperature. Fig. S2 TEM and ED pattern of CA-HT and CA-Si4. Fig. S3 Al 2p and Si 2p XPS spectra of HT and silicate anion intercalated HT materials. Table S1. Elemental composition of HT and silicate anion intercalated materials calculated using XRF spectrometer. See DOI: 10.1039/c3ra46703a |
| This journal is © The Royal Society of Chemistry 2014 |