Monobromomalononitrile: an efficient regioselective mono brominating agent towards active methylene compounds and enamines under mild conditions†
Sudipta Pathak,
Ashis Kundu and
Animesh Pramanik*
Department of Chemistry, University of Calcutta, 92, A. P. C. Road, Kolkata-700 009, India. E-mail: animesh_in2001@yahoo.co.in; Fax: +91-33-2351-9755; Tel: +91-33-2484-1647
First published on 31st January 2014
Abstract
The potential of monobromomalononitrile (MBM) as a convenient source of cationic bromine in organic bromination reaction has been explored. Studies reveal that MBM can be a good substitute for N-bromosuccinimide (NBS) in various respects. Enamines and active methylene compounds bearing aromatic rings are selectively mono brominated on the vinylic and active methylene group respectively on reaction with MBM. This methodology has the advantages of easy preparation of MBM, shorter reaction time and high yields of the product formation. Moreover it provides a metal free green brominating agent which is more convenient for the pharmaceutical industry. Mono bromination reaction takes place only on active methylene groups even after addition of excess amount of MBM. Enamines containing electron withdrawing, electron donating and ortho substituted amines react smoothly affording only the vinylic mono bromo products in good yields without producing any side products.
Bromination on organic molecules has been a workhorse in the field of organic synthesis because of the commercial significance of intermediate bromoorganics for the construction of important natural products, as well as in the production of intermediates for agrochemicals and pharmaceuticals. As for example, a huge number of commercially important products such as herbicides, pesticides, fire retardants, and other new important materials have bromo functionality.1 Formation of C–C bond using cross-coupling reactions can be employed with these bromides. Therefore, the halide compounds have huge impact on Heck,2 Stille–Suzuki3 and Sonogashira4 and other hetero coupling reaction via aromatic functionalization process.5 The use of elemental bromine is the traditional method of bromination. Careful control of the temperature and rate of bromine addition to avoid undesirable side reactions are the main disadvantages for the use of elemental bromination.6 Moreover, corrosive and toxic nature of elemental bromine is a serious drawback for handling elemental bromine. Therefore, milder brominating agents are urgently needed for the bromination on organic molecules.7–13 Various solid organic ammonium tribromides like 2,4-diamino-1,3-thiazole hydrotribromide,8b Bu4NBr3,8d,e PyHBr3,8e–g 1,2-dipyridiniumditribromide-ethane (DPTBE),8h Me4NBr3,8a 1,8-diazabicyclo[5.4.0]undec-7-ene hydrobromide perbromide (DBUHBr3),8c and phenyltrimethylammonium tribromide8e are employed for bromination avoiding the direct use of molecular bromine. Another method involves oxidative bromination using hydrogen bromide and bromide salts as a bromine source where bromine is generated in situ in the reaction mixture upon oxidation of bromide ions.9–13 The mostly employed bromides and oxidants combinations are H2O2–V2O5–Et4NBr,9d oxone/HBr,10b oxone (the active component is potassium monopersulfate, KHSO5)/NaBr,10a H2O2–HBr,9a–c t-BuOOH–HBr,9b,c Selectfluor®/KBr,12 NaBrO3–NaBr,13 CAN/LiBr,11b and cerium(IV) ammonium nitrate (CAN)/KBr.11a In all these reactions the use of binary solvent systems (an organic solvent and water) is necessary for satisfactory result. The most significant drawback of all these methods is the undesirable oxidation of the sensitive functional groups present in the substrates by the oxidizing agents.
With respect to the above mentioned methodologies, N-bromosuccinimide (NBS) is one of the most potent brominating agents due to its stability and safe and easy handling.14 NBS can brominate efficiently the activated aromatic compounds like phenols, amines etc.14d,e It can also brominate double bonds to 1,2-dibromo compounds; carbonyl compounds to α-brominated compounds;14c active methylene compounds to mon- and di-brominated compounds14f and enamines to vinylic and allylic brominated compounds.14g Though it is a mild brominating agent, it gives various side products on bromination of aromatic compounds14d,e and enamines or active methylene group.14c,g So it is a less selective brominating agent. For bromination of enamines containing electron rich aromatic ring, the selectivity of NBS is lost as it may brominate both the active aromatic ring and enamines, moreover the latter may also give rise to a mixture of vinylic and allylic brominated compounds. Herein we wish to report a general, efficient and highly selective brominating agent for mono bromination of active methylene compounds and enamines under mild reaction condition.
Monobromomalononitrile (MBM)15 is reported in the literature for the syntheses of various important compounds where it participates mainly in addition reaction with carbonyl group,15a–c α,β-unsaturated double bond15a–c and isolated double bond.15d It is also used for synthesis of biologically important heterocycles.15e In all the cases malononitrile part of MBM is added in the products. To the best of our knowledge, the appropriate reaction condition is not explored as yet for MBM where it may act solely as a brominating agent. Therefore initially an optimization study is carried out with a model reaction between acetylacetone and MBM in various non polar solvents to examine whether it gives only the brominated product or the products from the usual addition reaction with carbonyl group and O or C-alkylation reaction of enol substrate. When the reaction is carried out in non polar solvents like CCl4, hexane, benzene and toluene employing 1.2 equivalent of MBM at room temperature, the reaction does not proceed at all (Table 1, entries 1–5). Literature results show that polar solvent is necessary to carry out the bromination reaction with NBS on double bond or active methyelene group. Since MBM is chemically analogous to NBS, various polar solvents are chosen for bromination (Table 1, entries 6–12). When polar protic solvent methanol is used as a reaction medium, TLC analysis suggests the formation of only one product along with substantial amount of unreacted starting material (Table 1, entry 6). The structure of the isolated product (yield ∼ 30%) is confirmed by IR, 1H NMR and 13C NMR spectroscopy and elemental analysis, which establishes the formation of only the mono brominated product 3-bromopentane-2,4-dione 2a (Table 2, entry 1). This result incites us to perform the reaction in various polar solvents of varying polarities. However when the reaction is carried out in ethanol or water, the yield of the product 2a is only 32 and 25% respectively indicating the unsuitability of protic polar solvent for the reaction (Table 1, entries 7 and 8). Intriguingly, the yield of the product 2a increases substantially in aprotic polar solvents in presence of 1.2 equivalent of MBM at room temperature (Table 1, entries 9–11). Moreover when the polarity of the employed aprotic solvents increases in the order THF, EtOAc and CH3CN the yield of 2a also increases from 45% to 70% (Table 1, entries 9–11). Gratifyingly, the maximum yield of the product 2a is obtained in aprotic polar solvent DMF, nearly 91%, employing 1.2 equivalent of MBM at room temperature (Table 1, entry 12).
| Entry | Solvent (10 ml) | Time (h) | Yield (%) |
|---|---|---|---|
| 1 | CCl4 | 2 | __ |
| 2 | CHCl3 | 2 | __ |
| 3 | Hexane | 2 | __ |
| 4 | Benzene | 2 | __ |
| 5 | Toluene | 2 | __ |
| 6 | Methanol | 2 | 30 |
| 7 | Ethanol | 2 | 32 |
| 8 | H2O | 2 | 25 |
| 9 | Tetrahydrofuran | 2 | 45 |
| 10 | Ethyl acetate | 2 | 49 |
| 11 | Acetonitrile | 2 | 70 |
| 12 | DMF | 0.5 | 91 |
| Entry | Substrates (1) | Products (2) | Time (min) | Yield (%) | Melting point/ref. 16 |
|---|---|---|---|---|---|
| 1 | 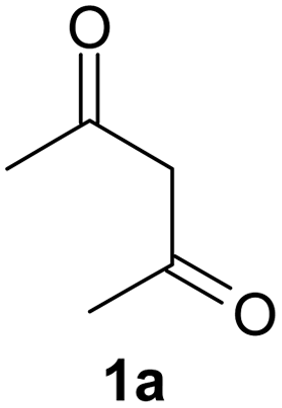 |
 |
30 | 91 | 137–139/138–140 |
| 2 |  |
 |
25 | 94 | 168–170/168–170 |
| 3 | 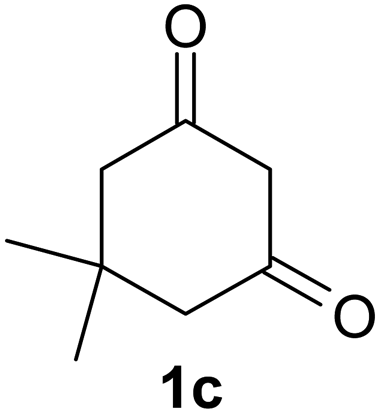 |
 |
25 | 93 | 172–176/174–176 |
| 4 |  |
 |
35 | 89 | 130–132/129 |
| 5 |  |
 |
40 | 86 | 195–197/196 |
| 6 |  |
 |
30 | 90 | 115–117/115–118 |
| 7 |  |
 |
25 | 86 | 180–182/182 |
| 8 | 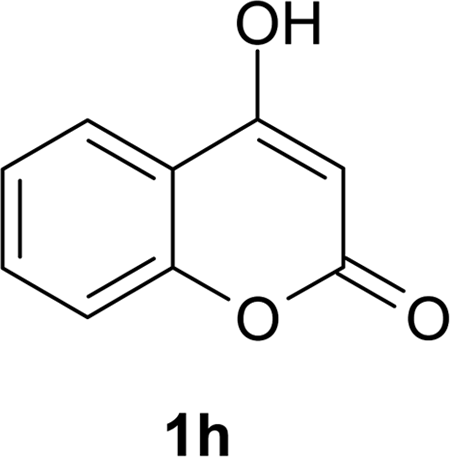 |
 |
35 | 93 | 190–192/192–194 |
After having prepared 2a successfully, we decide to explore the scope and generality of this reaction with various 1,3-dicarbonyl compounds including 1,3-cyclohexanedione (1b), 5,5-dimethylcyclohexane-1,3-dione (1c), acetoacetanilide (1d), barbituric acid (1e) and 1,3-indandione (1f) to furnish expected mono bromo-compound 2 in the optimized reaction conditions (Scheme 1 and Table 2). The results show that the reactions can produce the mono brominated products 2a–f in high yields within 25–40 min at room temperature (Table 2). This reaction is applicable to both cyclic as well as acyclic active methylene compounds. It is interesting to note that in presence of aromatic ring, bromination takes place only on active methylene group (1d) even after addition of excess amount of MBM (2.2 equiv.), confirmed by single crystal X-ray diffraction study (Fig. 1). Even the presence of excess amount of MBM (2.2 equiv.), cannot produce α,α-dibromo derivatives. The reaction stops selectively at mono bromo stage. This result establishes that the reaction of MBM is very specific for the formation of mono bromo derivatives of 1,3-dicarbonyl compounds. Activated compounds like 5-methyl-2H-pyrazole-3-ol (1g) and 4-hydroxycoumarin (1h) are also brominated with MBM to produce 4-bromo-5-methyl-2H-pyrazole-3-ol (2g) and 3-bromo-4-hydroxycoumarin (2h) respectively in high yields within 30 min at room temperature (Table 2, entries 7 and 8).
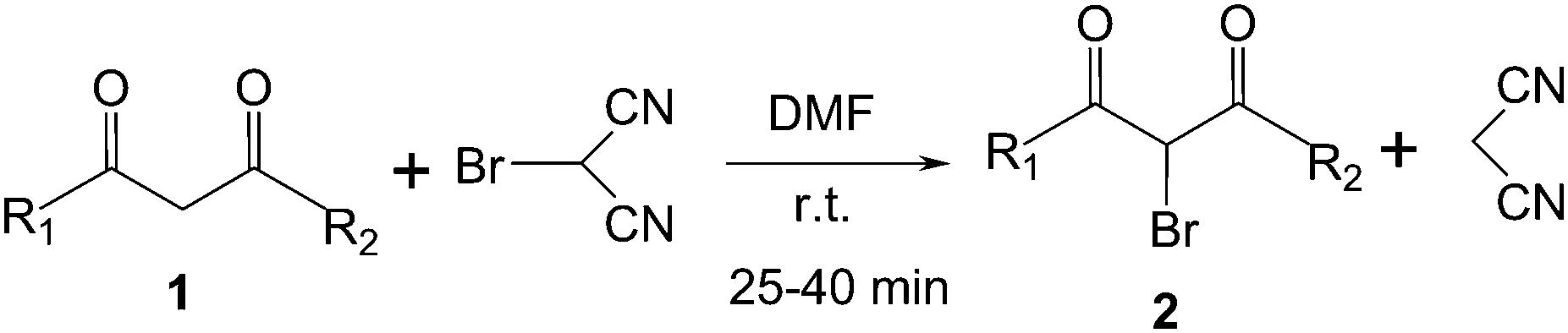 | ||
| Scheme 1 Bromination on active methylene group using MBM. | ||
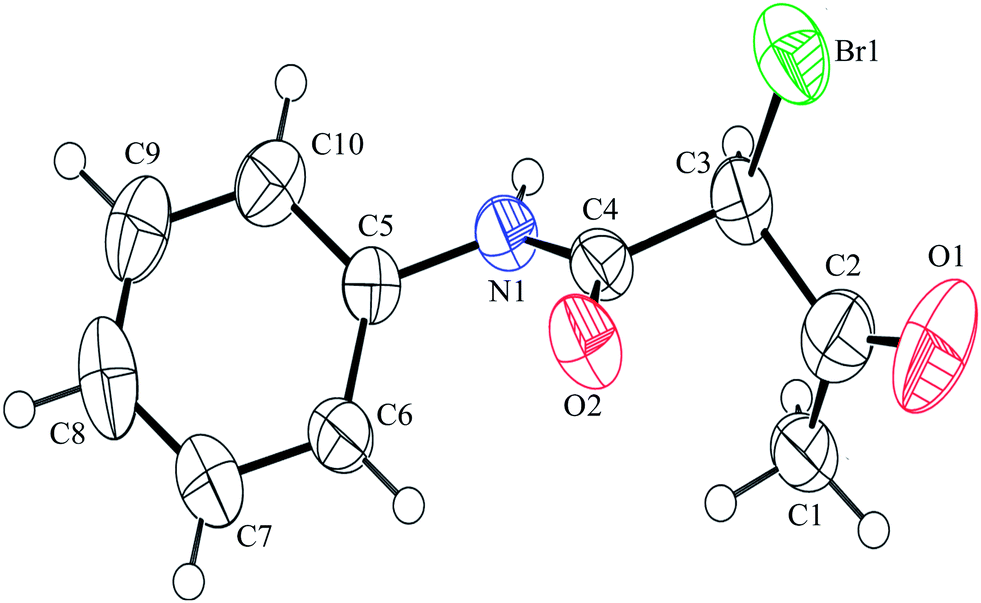 | ||
| Fig. 1 The crystal structure of 2d, the mono brominated product of acetoacetanilide (1d). | ||
Subsequently we explore the brominating ability of MBM with various substituted phenols (phenol, m-cresol, o-cresol, p-cresol, p-methoxyphenol, o-chlorophenol and m-aminophenol), anilines (aniline, m-anicidine, o-chloraniline and p-fluorophenol), methylketone carbonyl compounds (acetophenone, p-chloroacetophenone, p-nitroacetophenone and p-methoxyacetophenone), alkenes (styrene) and alkynes (phenylacetylene). But MBM does not react with all these substrates even when the reactions are carried out at high temperature using excess amount of MBM (2.2 equivalent) and allowing a prolonged reaction time. It is interesting to note that even activated aromatic ring like m-aminophenol does not produce any brominated product. Since phenoxide ion is more reactive than phenol, the above reaction has also been carried out in basic medium employing aqueous sodium hydroxide and organic base triethyl amine separately. But in both the cases the brominated compounds are not formed. In fact MBM is destroyed in aqueous sodium hydroxide solution. The results demonstrate that MBM does not react with aromatic compounds even under drastic condition.
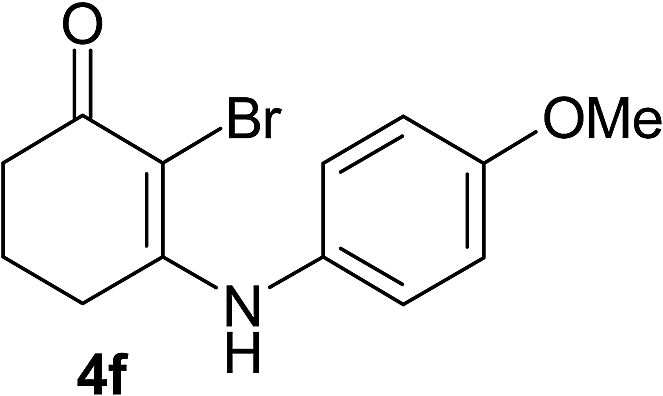
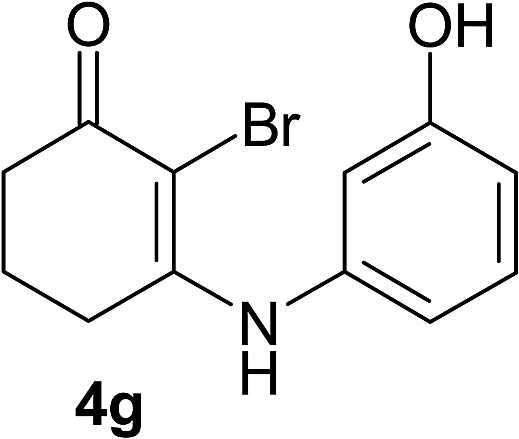
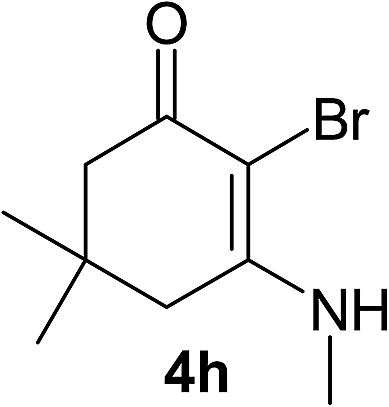
There are several reports of bromination of enamino compounds by using Br2/CCl4, NBS/MeOH, BrCN and NBS/montmorillonite (K-10).17 All these procedures give low to moderate yield of the vinylic brominated enamines after prolonged reaction time.17a,c Moreover some of these methods have serious drawbacks of formation of side products like allylic brominated enamines.17a,b But when the enamine of 1,3-cyclohexandione and benzylamine is treated with MBM in DMF at room temperature, only the vinylic brominated enamine is formed in good yield (Scheme 2 and Table 3). Then this simple procedure has been applied to different enamines of 1,3-cyclohexandione and dimedone. As evident from Table 3, several enamines 3 containing electron withdrawing and electron donating amines react smoothly with MBM affording the vinylic mono bromo products 4 in good yields without producing any side products. The enamines 3f, 3g, 3o and 3p although contain electronically rich aromatic ring; bromination takes place only at the vinylic position. Even in presence of ortho substituted amine as in the case of enamines 3e and 3n, the formation of vinylic brominated product is not hindered due to the steric reason. The structures of all the mono brominated products were determined by matching the reported melting points and also the spectroscopic data. Furthermore, the formation of product 4a is confirmed by X-ray crystallographic analysis (Fig. 2).
 | ||
| Scheme 2 Bromination on enamines of 1,3-cyclohexanedione and dimedone. | ||
| Entry | Substrate | Product | Time (min) | Yield (%) | Observed/ref. melting point17c (°C) |
|---|---|---|---|---|---|
| 1 |  |
 |
30 | 94 | 128–130 |
| 2 |  |
 |
35 | 92 | 96–98 |
| 3 |  |
 |
37 | 91 | 131–133 |
| 4 |  |
 |
40 | 89 | 122–124 |
| 5 |  |
 |
40 | 86 | 158–160 |
| 6 |  |
 |
28 | 92 | 140–142 |
| 7 | 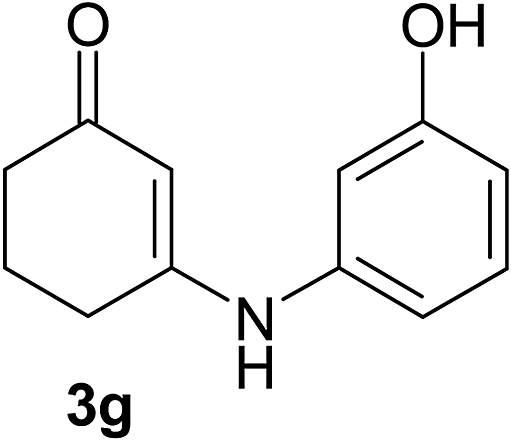 |
 |
25 | 91 | 176–178 |
| 8 |  |
 |
25 | 95 | 179–181/177–179 |
| 9 | 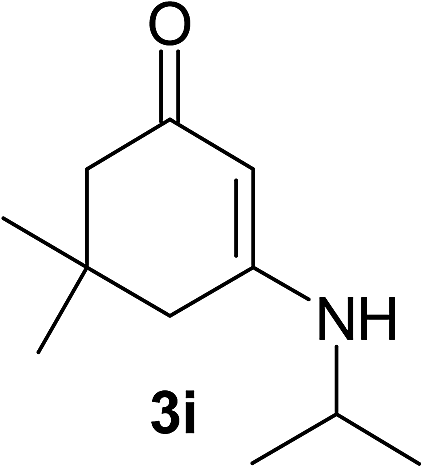 |
 |
25 | 94 | 89–91/90–92 |
| 10 |  |
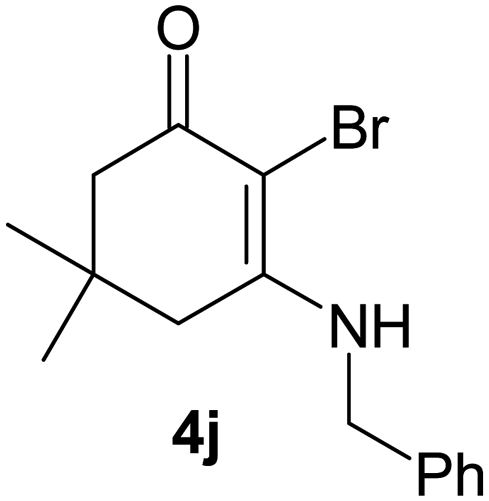 |
30 | 94 | 183–185/186–188 |
| 11 | 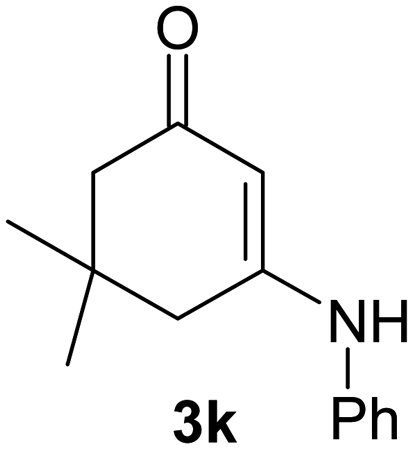 |
 |
36 | 90 | 156–158/158–159 |
| 12 |  |
 |
39 | 88 | 178–180 |
| 13 | 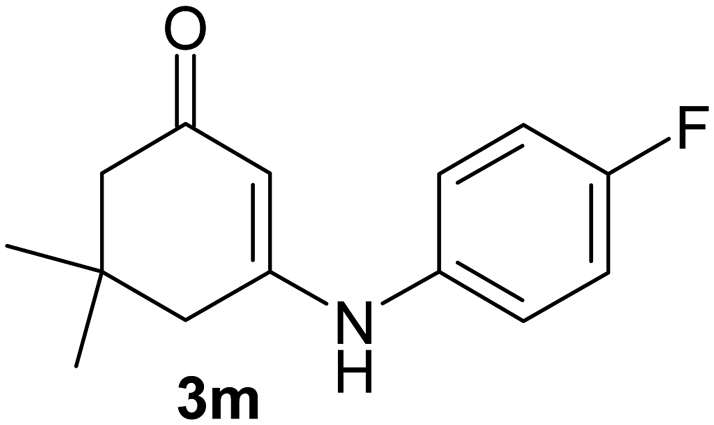 |
 |
42 | 86 | 156–158 |
| 14 | 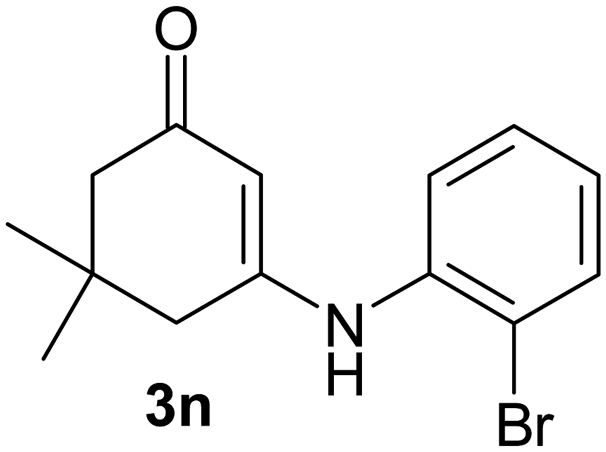 |
 |
45 | 85 | 146–148 |
| 15 | 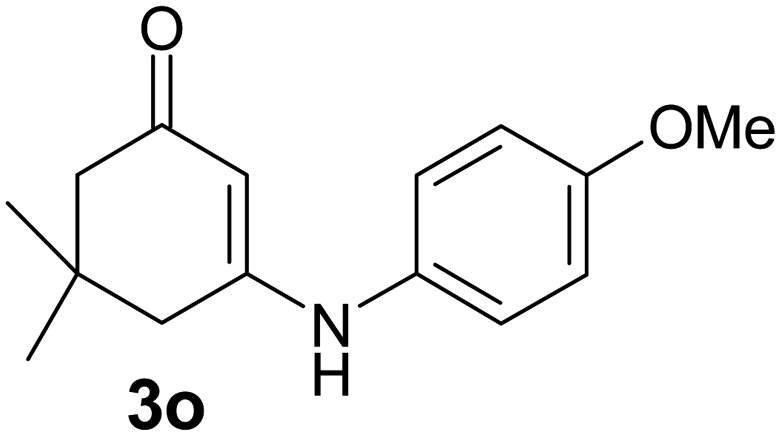 |
 |
30 | 90 | 128–130 |
| 16 |  |
 |
35 | 92 | 188–190 |
 | ||
| Fig. 2 The crystal structure of 4a, the mono brominated product of 3a. | ||
The formation of brominated compounds 4 from enamines 3 can be explained on the basis of the proposed mechanism depicted in Scheme 3.18 At first, the activated double bond of enamine attacks the electropositive bromine of MBM to generate bromo intermediate 5 and malononitrate anion. Finally malononitrate anion abstracts proton from cationic intermediate 5 to form mono bromo emino derivatives 4. Since the reaction passes through an ionic path way, a polar medium is necessary for the reaction. The poor product formation in protic polar solvents e.g. methanol, ethanol and water may be due to the protonation of malononitrate anion from solvents (Table 1, entries 6–8).
 | ||
| Scheme 3 Plausible mechanism for bromination of enamines 3 by MBM. | ||
In order to assess the relative efficiency and selectivity of NBS and MBM in mono bromination of active methylene compounds and enamines some representative studies have been carried out under similar reaction conditions (Table 4). In case of NBS the formation of mixture of products is observed by TLC and NMR analysis and the results are included in Table 4. The results clearly demonstrate that NBS is less selective in bromination producing mixture of brominated products. On the other hand MBM produces only mono brominated product without formation of any side products. Therefore MBM is a superior mono brominating agent towards active methylene compounds and enamines.19
| Substrate | MBMa | NBSa | ||
|---|---|---|---|---|
| Product | Yield (%) | Product | Yield (%) | |
| a Reactions are carried out in DMF medium at room temperature with 1.2 equivalent brominating agent with respect to substrate.b Mixture of products obtained. | ||||
| 1a | 2a | 91 | 2a | 91 |
| 1b | 2b | 94 | 2bb | 65 |
| 1c | 2c | 93 | 2cb | 74 |
| 1d | 2d | 89 | 2db | 53 |
| 1e | 2e | 86 | 2eb | 66 |
| 1f | 2f | 90 | 2fb | 69 |
| 1g | 2g | 86 | 2gb | 75 |
| 1h | 2h | 93 | 2h | 89 |
| 3b | 4b | 92 | 4b | 89 |
| 3c | 4c | 91 | 4c | 90 |
| 3e | 4e | 86 | 4e | 80 |
| 3f | 4f | 92 | 4fb | 80 |
| 3g | 4g | 91 | 4gb | 73 |
| 3k | 4k | 90 | 4k | 89 |
| 3l | 4l | 88 | 4l | 85 |
| 3n | 4n | 85 | 4n | 75 |
| 3o | 4o | 90 | 4ob | 60 |
| 3p | 4p | 92 | 4pb | 60 |
In conclusion, we have successfully developed a set of mild reaction conditions for MBM where it can act as a selective and efficient mono brominating agent. The efficacy of the methodology lies in the bromination of 1,3-dicarbonyl compounds and enamines containing activated aromatic rings. This methodology has the advantages of easy preparation of MBM, shorter reaction time and high yields of the product formation. The less reactive MBM can be a very good substitute for relatively more reactive NBS in regioselective mono bromination of 1,3-dicarbonyl compounds and enamines. Moreover the application of this metal free organo brominating agent is environmental friendly and can be considered as a green reagent within the domain of Green Chemistry principles.
Experimental section
General information
Starting materials and solvents are purchased from commercial suppliers and used without further purification. Melting points are determined in open capillary tubes and are uncorrected. IR spectra are recorded on a Perkin-Elmer 782 spectrophotometer. 1H (300 MHz) and 13C NMR (75 MHz) spectra are recorded on Bruker 300 MHz instrument in [D6]DMSO or in CDCl3. Elemental analyses (C, H and N) are performed using Perkin-Elmer 240C elemental analyzer. The X-ray diffraction data for crystallized compounds are collected with MoKα radiation at 296 K using the Bruker APEX-II CCD System. The crystals are positioned at 50 mm from the CCD. Frames are measured with a counting time of 5 s. Data analyses are carried out with the Bruker APEX2 and Bruker SAINT program. The structures are solved using direct methods with the Shelxs 97 program (Sheldrick, 2008).General procedure for the preparation of monobromomalononitrile (MBM)
A solution of malononitrile (3.3 g, 0.05 mol) in water (10 ml) and 2-propanol (10 ml) was cooled in an ice cold water-bath at 20 °C. Then bromine (8.0 g, 0.05 mol) was added slowly into the solution with constant stirring, during which the temperature of the reaction mixture will rise and the colour of bromine will disappear immediately. After the addition of bromine, the reaction mixture was stirred at room temperature for 15 min and left at 0 °C for 10 h. The solid monobromomalononitrile was precipitated out from the reaction mixture and filtered off through suction.General procedure for the preparation of 2a–h and 4a–p
The compounds 1a–h or 3a–p (1.0 mmol) were dissolved in dimethylformamide (5 ml) at room temperature. Then monobromomalononitrile (00.174 g, 1.2 mmol) was added and the resulting mixture was stirred at room temperature for 25–45 min (as mentioned in Table 2). Then the reaction mixture was poured into cold water and extracted with ethylacetate. Ethylacetate was removed under reduced pressure to get a gummy mass which was purified by column chromatography on silica gel using ethylacetate/hexane to obtain pure 2a–h and 4a–p.2-Bromo-3-oxo-N-phenylbutyramide (2d): IR (KBr) 3238, 1738 cm−1; 1H NMR (300 MHz, CDCl3) δH 8.44 (br s, 1H), 7.52 (d, J = 7.5 Hz, 2H), 7.35 (t, J = 8.4 Hz, 2H), 7.17 (t, J = 6.3 Hz, 1H), 4.88 (s, 1H), 2.48 (s, 3H); 13C NMR (75 MHz, CDCl3) δC 198.1, 161.7, 136.7, 129.1, 125.4, 120.1, 49.5, 27.3.
3-Benzylamino-2-bromocyclohex-2-enone (4a): IR (KBr) 3180, 1570 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.42–7.24 (m, 5H), 6.08 (br s, 1H), 4.52 (d, J = 6 Hz, 2H), 2.59–2.48 (m, 4H), 2.00–1.94 (m, 2H); 13C NMR (75 MHz, CDCl3) δC 187.8, 161.1, 137.0, 129.1, 128.0, 126.7, 96.5, 47.3, 36.7, 26.7, 20.8.
2-Bromo-3-phenylaminocyclohex-2-enone (4b): IR (KBr) 3195, 1590 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.43–7.27 (m, 3H), 7.16 (d, J = 7.5 Hz, 2H), 2.59–2.57 (m, 4H), 196–1.92 (m, 2H); 13C NMR (75 MHz, CDCl3) δC 188.5, 159.4, 137.3, 129.5, 126.9, 125.8, 97.9, 37.2, 28.2, 21.4.
2-Bromo-3-(4-chlorophenylamino)-cyclohex-2-enone (4c): IR (KBr) 3202, 1610 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.39–7.28 (m, 2H), 7.12–7.10 (m, 2H), 2.59–2.47 (m, 4H), 1.99–1.94 (m, 2H); 13C NMR (75 MHz, CDCl3) δC 188.56, 158.81, 135.8, 132.5, 129.5, 126.9, 98.4, 37.0, 28.0, 21.3.
2-Bromo-3-(4-fluorophenylamino)-cyclohex-2-enone (4d): IR (KBr) 3233, 1633 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.30 (d, J = 1.8 Hz, 1H), 7.21–7.08 (m, 4H), 2.57–2.48 (m, 4H), 1.99–1.91 (m, 2H); 13C NMR (75 MHz, CDCl3) δC 188.6, 163.0, 159.7, 133.3, 128.2, 128.1, 116.5, 116.2, 97.8, 37.1, 28.1, 21.3.
2-Bromo-3-(4-bromophenylamino)-cyclohex-2-enone (4e): IR (KBr) 3185, 1590 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.67 (d, J = 7.8 Hz, 1H), 7.48–7.16 (m, 3H), 2.60–2.49 (m, 4H), 2.02–1.96 (m, 2H); 13C NMR (75 MHz, CDCl3) δC 188.8, 158.8, 136.2, 133.5, 128.3, 128.3, 127.5, 121.2, 99.1, 37.1, 28.0, 21.4.
2-Bromo-3-(4-methoxyphenylamino)-cyclohex-2-enone (4f): IR (KBr) 3196, 1601 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.28 (br s, 1H), 7.10 (d, J = 8.7 Hz, 2H), 6.92 (d, J = 9 Hz, 2H), 3.85 (s, 3H), 2.5 (t, J = 6.3 Hz, 2H), 2.47 (t, J = 6 Hz, 2H), 1.96–1.88 (m, 2H); 13C NMR (75 MHz, CDCl3) δC 188.5, 160.3, 158.7, 129.9, 127.8, 114.6, 97.0, 55.5, 37.1, 28.1, 21.3.
2-Bromo-3-(4-hydroxyphenylamino)-cyclohex-2-enone (4g): IR (KBr) 3350, 3215, 1575 cm−1; 1H NMR (300 MHz, D6-DMSO) δH 9.62 (s, 1H), 8.56 (s, 1H), 7.16 (t, J = 7.8 Hz, 1H), 6.68–6.61 (m, 3H), 2.50–2.34 (m, 4H), 1.80–1.78 (m, 2H); 13C NMR (75 MHz, D6-DMSO) δC 187.6, 160.8, 158.2, 139.6, 130.0, 117.4, 113.8, 113.8, 96.2, 37.4, 29.0, 21.7.
2-Bromo-3-(4-chlorophenylamino)-5,5-dimethylcyclohex-2-enone (4l): IR (KBr) 3185, 1572 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.39 (d, J = 8.4 Hz, 2H), 7.28 (br s, 1H), 7.10 (d, J = 8.7 Hz, 2H), 2.43 (s, 2H), 2.38 (s, 2H), 1.04 (s, 6H); 13C NMR (75 MHz, CDCl3) δC 188.2, 156.8, 135.9, 132.5, 129.6, 127.0, 97.3, 50.6, 41.4, 32.8, 27.9.
2-Bromo-3-(4-fluorophenylamino)-5,5-dimethylcyclohex-2-enone (4m): IR (KBr) 3205, 1622 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.27–7.14 (m, 4H), 2.42 (s, 2H), 2.23 (s, 2H), 1.04 (s, 6H); 13C NMR (75 MHz, CDCl3) δC 188.1, 163.0, 157.4, 133.3, 128.1, 128.0, 116.6, 116.3, 96.7, 50.7, 41.4, 32.7, 27.9.
2-Bromo-3-(2-bromophenylamino)-5,5-dimethylcyclohex-2-enone (4n): IR (KBr) 3201, 1615 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.69 (d, J = 7.8 Hz, 1H), 7.36 (t, J = 7.5 Hz, 1H), 7.26–7.21 (m, 2H), 2.44 (s, 2H), 2.31 (s, 2H), 1.05 (s, 6H); 13C NMR (75 MHz, CDCl3) δC 188.3, 156.8, 136.3, 133.5, 128.4, 128.3, 127.9, 121.6, 97.3, 50.7, 41.3, 32.7, 28.0.
2-Bromo-3-(4-methoxyphenylamino)-5,5-dimethylcyclohex-2-enone (4o): IR (KBr) 3212, 1607 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.27 (br s, 1H), 7.08 (d, J = 7.2 Hz, 2H), 6.90 (d, J = 7.2 Hz, 2H), 3.87 (s, 3H), 2.42 (s, 2H), 2.32 (s, 2H), 0.96 (s, 6H); 13C NMR (75 MHz, CDCl3) δC 188.0, 158.7, 158.2, 130.0, 127.9, 114.7, 95.9, 55.5, 50.7, 41.4, 32.5, 28.0.
2-Bromo-3-(3-hydroxyphenylamino)-5,5-dimethylcyclohex-2-enone (4p): IR (KBr) 3330, 3217, 1601 cm−1; 1H NMR (300 MHz, D6-DMSO) δH 9.65 (br s, 1H), 8.60 (br s, 1H), 7.18 (t, J = 6.6 Hz, 1H), 6.92–6.60 (m, 3H), 2.43 (s, 2H), 2.29 (s, 2H), 0.93 (s, 6H); 13C NMR (75 MHz, D6-DMSO) δC 187.2, 158.6, 158.3, 139.6, 130.1, 117.4, 113.8, 113.1, 95.1, 50.8, 42.0, 32.8, 27.7.
Acknowledgements
S.P. and A.K. thank CSIR and UGC New Delhi, India, for offering Senior Research Fellowship respectively. The financial assistance of CSIR, New Delhi is gratefully acknowledged [Major Research Project, no. 02(0007)/11/EMR-II]. Crystallography was performed at the DST-FIST, India-funded Single Crystal Diffractometer Facility at the Department of Chemistry, University of Calcutta.References
- (a) G. W. Gribble, Chem. Soc. Rev., 1999, 28, 335 RSC; (b) A. Butler and J. V. Walker, Chem. Rev., 1993, 93, 1937 CrossRef CAS; (c) R. H. Seevers and R. E. Counsell, Chem. Rev., 1982, 82, 575 CrossRef CAS.
- (a) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed; (b) W. Cabri and I. Candiani, Acc. Chem. Res., 1995, 28, 2 CrossRef CAS; (c) A. Meijere and F. E. Meyer, Angew. Chem., Int. Ed., 1995, 33, 2379 CrossRef; (d) M. R. Eberhard, Org. Lett., 2004, 6, 2125 CrossRef CAS PubMed; (e) M. Oestreich, F. Sempere-Culler and A. B. Machotta, Angew. Chem., Int. Ed., 2004, 44, 149 CrossRef PubMed; (f) C. J. Kressierer and T. J. J. Müller, Angew. Chem., Int. Ed., 2004, 43, 5997 CrossRef CAS PubMed; (g) J.-C. Xiao, B. Twamley and J. M. Shreeve, Org. Lett., 2004, 6, 3845 CrossRef CAS PubMed; (h) Q. Yao, E. P. Kinney and C. Zheng, Org. Lett., 2004, 6, 2997 CrossRef CAS PubMed; (i) L. A. Arnold, W. Luo and R. K. Guy, Org. Lett., 2004, 6, 3005 CrossRef CAS PubMed; (j) S. Braese and A. de Meijere, Metal-Catalyzed Cross-Coupling Reactions, ed. A. De Meijere and F. Diederich, Wiley-VCH, Weinheim, Germany, 2004, pp. 217–315 Search PubMed.
- (a) J. K. Stille, Pure Appl. Chem., 1985, 57, 1771 CrossRef CAS; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (c) W. A. Herrmann, C.-P. Reisinger and P. Haerter, Aqueous-Phase Organometallic Catalysis, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, Germany, 2004, pp. 511–523 Search PubMed; (d) D. Zhao, Z. Fei, T. J. Geldbach, R. Scopelliti and P. J. Dyson, J. Am. Chem. Soc., 2004, 126, 15876 CrossRef CAS PubMed; (e) A. C. Spivey, C. J. G. Gripton and J. P. Hannah, Curr. Org. Synth., 2004, 1, 211 CrossRef CAS; (f) R. B. Bedford, S. L. Hazelwood, M. E. Limmert, D. A. Albisson, S. M. Draper, P. N. Scully, S. J. Coles and M. B. Hursthouse, Chem. – Eur. J., 2003, 9, 3216 CrossRef CAS PubMed; (g) B. M. Choudary, M. S. Chowdari, S. Naidu, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2002, 124, 14127 CrossRef CAS PubMed.
- (a) K. Sonogashira, Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, New York, 1991, vol. 3, p. 521 Search PubMed; (b) K. Sonogashira, Metal-Catalyzed Cross-Coupling Reactions, F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, Germany, 1998; p. 203 Search PubMed; (c) D. García, A. M. Cuadro, J. Alvarez-Builla and J. J. Vaquero, Org. Lett., 2004, 6, 4175 CrossRef PubMed; (d) R. B. De Vashre, L. R. Moore and K. H. Shaughnessy, J. Org. Chem., 2004, 69, 7919 CrossRef PubMed; (e) S. Urgaonkar and J. G. Verkade, J. Org. Chem., 2004, 69, 5752 CrossRef CAS PubMed; (f) J. Cheng, Y. Sun, F. Wang, M. Guo, J.-H. Xu, Y. Pan and Z. Zhang, J. Org. Chem., 2004, 69, 5428 CrossRef CAS PubMed.
- (a) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046 CrossRef CAS; (b) J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805 CrossRef CAS; (c) J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852 CrossRef CAS.
- (a) P. B. De la Mare, Electrophilic Halogenation, Cambridge University Press, Cambridge, UK, 1976, ch. 5 Search PubMed; (b) R. Taylor, Electrophilic Aromatic Substitution, Wiley, Chichester, UK, 1990 Search PubMed.
- (a) M. Eissen and D. Lenoir, Chem. – Eur. J., 2008, 14, 9830 CrossRef CAS PubMed; (b) M. Eissen, M. Strudthoff, S. Backhaus, C. Eismann, G. Oetken, S. Kaling and D. Lenoir, J. Chem. Educ., 2011, 88, 284 CrossRef CAS; (c) V. V. K. M. Kandepi and N. Narender, Synthesis, 2012, 15 CAS; (d) H. Xue, H. Tan, D. Wei, Y. Wei, S. Lin, F. Liang and B. Zhao, RSC Adv., 2013, 3, 5382 RSC; (e) S. Gazi and R. Ananthakrishnan, RSC Adv., 2012, 2, 7781 RSC; (f) X.-L. Li, W. Wu, X.-H. Fan and L.-M. Yang, RSC Adv., 2013, 3, 12091 RSC; (g) Y. Nishina, J. Morita and B. Ohtani, RSC Adv., 2013, 3, 2158 RSC.
- (a) M. Avramoff, J. Weiss and O. Schächter, J. Org. Chem., 1963, 28, 3256 CrossRef CAS; (b) L. Forlani, Synthesis, 1980, 487 CrossRef CAS; (c) H. A. Muathen, J. Org. Chem., 1992, 57, 2740 CrossRef CAS; (d) M. K. Chaudhuri, A. T. Khan, B. K. Patel, D. Dey, W. Kharmawophlang, T. R. Lakshmiprabha and G. C. Mandal, Tetrahedron Lett., 1998, 39, 8163 CrossRef CAS; (e) K. Tanaka, R. Shiraishi and F. Toda, J. Chem. Soc., Perkin Trans. 1, 1999, 3069 RSC; (f) F. Toda and J. Schmeyers, Green Chem., 2003, 5, 701 RSC; (g) S. Nakamatsu, S. Toyota, W. Jones and F. Toda, Chem. Commun., 2005, 3808 RSC; (h) V. Kavala, S. Naik and B. K. Patel, J. Org. Chem., 2005, 70, 4267 CrossRef CAS PubMed.
- (a) T. L. Ho, B. Gupta and G. A. Olah, Synthesis, 1977, 676 CrossRef CAS; (b) N. B. Barhate, A. S. Gajare, R. D. Wakharkar and A. V. Bedekar, Tetrahedron Lett., 1998, 39, 6349 CrossRef CAS; (c) N. B. Barhate, A. S. Gajare, R. D. Wakharkar and A. V. Bedekar, Tetrahedron, 1999, 55, 11127 CrossRef CAS; (d) U. Bora, G. Bose, M. K. Chaudhuri, S. S. Dhar, R. Gopinath, A. T. Khan and B. K. Patel, Org. Lett., 2000, 2, 247 CrossRef CAS PubMed.
- (a) R. K. Dieter, L. E. Nice and S. E. Velu, Tetrahedron Lett., 1996, 37, 2377 CrossRef CAS; (b) K.-M. Kim and I.-H. Park, Synthesis, 2004, 2641 CrossRef CAS PubMed.
- (a) V. Nair, S. B. Panicker, A. Augustine, T. G. George, S. Thomas and M. Vairamani, Tetrahedron, 2001, 57, 7417 CrossRef CAS; (b) S. C. Roy, C. Guin, K. K. Rana and G. Maiti, Tetrahedron Lett., 2001, 42, 6941 CrossRef CAS.
- C. Ye and J. M. Shreeve, J. Org. Chem., 2004, 69, 8561 CrossRef CAS PubMed.
- A. Subbarayappa, S. Ghosh, P. U. Patoliya, G. Ramachandraiah, M. Agrawal, M. R. Gandhi, S. C. Upadhyay, P. K. Ghosh and B. C. Ranu, Green Chem., 2008, 10, 232 RSC.
- (a) C. K. Tan and Y.-Y. Yeung, Chem. Commun., 2013, 49, 7985 RSC; (b) M. S. Markoulides, C. P. Ioannou, M. J. Manos and N. Chronakis, RSC Adv., 2012, 2, 12269 RSC; (c) R. B. Mohan and N. C. G. Reddy, Synth. Commun., 2013, 43, 2603 CrossRef CAS; (d) K. Venkateswarlu, K. Suneel, B. Das, K. N. Reddy and T. S. Reddy, Synth. Commun., 2009, 39, 215 CrossRef CAS; (e) P. K. Chhattise, A. V. Ramaswamy and S. B. Waghmode, Tetrahedron Lett., 2008, 49, 189 CrossRef CAS PubMed; (f) A. Rahman and S. B. Jonnalagadda, Synth. Commun., 2012, 42, 1091 CrossRef CAS; (g) M. E. F. Braibante, H. T. S. Braibante, G. B. Rosso and J. K. da Roza, Synthesis, 2001, 1935 CrossRef CAS.
- (a) I. N. Bardasov, O. V. Kayukova, Y. S. Kayukov, O. V. Ershov, M. Y. Belikov and O. E. Nasakin, Chem. Heterocycl. Compd., 2009, 45, 1035 CrossRef CAS PubMed; (b) O. V. Kayukova, Y. S. Kayukov, E. S. Lapteva, I. N. Bardasov, O. V. Ershov and O. E. Nasakin, Russ. J. Org. Chem., 2006, 42, 1414 CrossRef CAS; (c) I. N. Bardasov, O. V. Kayukova, Y. S. Kayukov, O. V. Ershov and O. E. Nasakin, Russ. J. Appl. Chem., 2009, 82, 1431 CrossRef CAS; (d) Y. Kita, K. Gotanda, K. Murata, M. Suemura, A. Sano, T. Yamaguchi, M. Oka and M. Matsugi, Org. Process Res. Dev., 1998, 2, 250 CrossRef CAS; (e) A. Davoodnia, S. Ameli and N. Tavakoli-Hoseini, Asian J. Chem., 2011, 23, 3707 CAS.
- (a) R. Hosseinzadeh, M. Tajbakhsh, M. Mohadjerani and Z. Lasemi, Monatsh. Chem., 2009, 140, 57 CrossRef CAS PubMed; (b) H. R. Eisenhauer and K. P. Link, J. Am. Chem. Soc., 1954, 76, 1647 CrossRef CAS; (c) A. Arrieta, I. Ganboa and C. Palomo, Synth. Commun., 1984, 14, 939 CrossRef CAS; (d) K. D. S. Puri, S. Sood and A. Muthuraman, Med. Chem. Res., 2012, 21, 2300 CrossRef CAS.
- (a) I. Jirkovsky, Can. J. Chem., 1974, 52, 55 CrossRef CAS; (b) A. Alberola, C. Andreés, G. A. Ortega, R. Pedrosa and M. Vicente, Synth. Commun., 1986, 16, 1161 CrossRef CAS; (c) E. F. M. Braibante, H. T. S. Braibante, G. B. Rosso and J. K. da Roza, Synthesis, 2001, 1935 CrossRef.
- N. S. Zefirov and D. I. Makhonkov, Chem. Rev., 1982, 82, 615 CrossRef CAS.
- Y. Honda, S. Katayama, M. Kojima, T. Suzuki and K. Izawa, Org. Lett., 2002, 4, 447 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 962394 and 962395. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra46687f |
| This journal is © The Royal Society of Chemistry 2014 |