
Comparison of the gelation behaviour of N-substituted tetradecanamide amphiphiles in organic liquids: effect of hydrogen-bonding ability of the head-group
Abstract
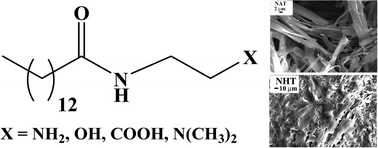
In this work, we have investigated the role of intermolecular hydrogen-bonding (H-bonding) interaction of the head-groups of amphiphilic organogelators. For this we have designed, synthesized and studied the gelation behaviour of a series of amide amphiphiles having hydrocarbon chains with the same length (C14) but with –COOH, –OH, –NH2 or –N(CH3)2) functionalities as head-group. These gelators, except the one with –N(CH3)2 head-group, efficiently gelated aromatic solvents. The gelation in all the solvents employed was observed to be thermoreversible. The gels were characterized by XRD spectroscopy, electron microscopy, and rheology. The amphiphiles were observed to form ribbon-like aggregates in aromatic solvents. The gel-to-sol transition temperatures as well as mechanical strengths of the organogels were observed to increase with the spacer length. The results suggest that the intermolecular H-bonding interactions between head-groups were essential for gelation.
 Please wait while we load your content...
Please wait while we load your content...

