Acid–chromic chloride functionalized natural clay-particles for enhanced conversion of one-pot cellulose to 5-hydroxymethylfurfural in ionic liquids
Yunlei Zhangab,
Jianming Pan*ab,
Mengying Ganab,
Hongxiang Oua,
Yongsheng Yanb,
Weidong Shib and
Longbao Yub
aSchool of the Environment and Safety Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: zhenjiangpjm@126.com
bSchool of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China
First published on 18th December 2013
Abstract
By grafting –SO3H and Cr(III) onto the surface of treated attapulgite (ATP) and halloysite nanotubes (HNTs), two acid–chromic chloride bi-functionalized catalysts i.e. ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) were successfully synthesized. Then the as-prepared catalysts were characterized by SEM, TEM, EDS, XPS, BET, NH3–TPD, nitrogen adsorption–desorption and TG analysis. The catalytic activities of the synthesized ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) were evaluated for the conversion of one-pot cellulose to 5-hydroxymethylfurfural (HMF) in an ionic liquid ([EMIM]-Cl) system. The amount of catalyst, reaction time and reaction temperature were optimized for cellulosic conversion over the two catalysts, resulting in a maximum yield of 31.20% for ATP–SO3H–Cr(III) and 41.22% for HNTs–SO3H–Cr(III) under the optimized conditions. The clay supported catalysts developed in this work showed greatly improved performances in cellulose-to-HMF conversion over other solid catalysts. In addition, the prepared catalysts could be very easily recycled, at least three times for ATP–SO3H–Cr(III) and twice for HNTs–SO3H–Cr(III) without significant loss of activity.
1. Introduction
With the seriousness of the energy crisis situation, it is time to find alternative resources in the chemical research field. Biomass-derived energy, especially using cellulose, which exists abundantly in nature and is composed of glucose units bonded via β-1,4-glucosidic bonds, has attracted a great deal of attention because of its renewable advantages. Biomass-derived products can be transformed into small molecules, such as glucose,1 5-hydroxymethylfurfural (HMF),2,3 and ethanol, which are regarded as biofuels.4,5 So far, much effort has been devoted to the generation of HMF from cellulose using efficient catalytic systems as it is an important intermediate for many useful fuels and chemicals. For example, 2,5-dimethylfuran (DMF), a transportation fuel that has a higher energy density than ethanol, can be formed by hydrogenation of HMF.6,7 Other chemicals such as 2,5-bis(hydromethyl) furan (2,5-BHF) and 2,5-diformylfuran (2,5-DFF), which are widely used in the synthesis of polymers, can also be produced from HMF.8 However, cellulose can not be dissolved in commonly used solvents such as H2O, ethyl acetate, acetonitrile and ether,9 which eliminates its wide-scale application. Recently, ionic liquids (ILs) which are designated as “green” solvents due to their extremely low vapor pressures, nonflammable nature, and thermal and chemical stability have emerged in this field, and have successfully solved the problem concerning the degradation of cellulose in many studies. What is more, ILs consist only of cations and anions, and many of them can be, in principle, indefinitely recycled in the process.10 So far, some types of ILs like 1-butyl-3-methylimidazolium chloride ([BMIM]-Cl), 1-ethyl-3-methylimidazolium chloride ([EMIM]-Cl), 1-carboxyethyl-3-methylimidazolium chloride ([C1COOHmim]-Cl), 1-butyl-3-methylimidazolium hydrogensulfate ([Bmim]HSO4),11,12 1-butyl sulfonic acid-3-methylimidazolium hydrogensulfate ([C4SO3Hmim]HSO4) 1-butyl sulfonic acid-3-methylimidazolium chloride ([C4SO3Hmim]-Cl),13,14 have been used as solvents to hydrolyze different kinds of lignocellulose (corn stalk, straw, pine wood and bagasse) to HMF, obtaining good yields.Seri reported the direct production of HMF from cellulose catalyzed by LaCl3 in water, but HMF was obtained in a low yield of 19% at 250 °C.15 Hussein's group used CrCl3 and CrCl3/CuCl2 catalysts in 1-ethyl-3-methylimidazolium chloride ([EMIM]-Cl) ionic liquid to degrade cellulose directly to HMF, and the yields of HMF were 35.6% and 39.9%, respectively.16 Their work also proved that CrCl3 was one of the most effective catalysts for the selective conversion of cellulose to HMF, with lower concentrations of other byproducts. Enlightened by the above information, CrCl3 was incorporated into 1-(3-sulfonic acid)propane-3-methylimidazole hydrosulfate ([PSMIM]HSO4) in a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by Zhou's group, and they successfully synthesized the functionalized IL Cr([PSMIM]HSO4)3 and got a perfect yield of HMF.17 Recent studies have demonstrated that the use of homogeneous media such as ionic liquids, may be beneficial for the catalytic conversion of cellulose. Compared to homogeneous systems, heterogeneous systems are easier to separate and recycle, and they have shown superior behavior in terms of yields of HMF. Liu et al. successfully prepared a novel and efficient mesoporous polydivinylbenzene (PDVB) based solid strong acid (PDVB–SO3H–SO2CF3) as a catalyst for biomass transformation.18 Bootsma used propylsulfonic acid and arenesulfonic acid functionalized silicas as organic–inorganic hybrid mesoporous catalysts for cellulose hydrolysis and these were found to catalyze the hydrolysis of cellobiose efficiently, with apparent activation energies similar to those reported previously for homogeneous organic acids.19 The solid catalysts presented excellent catalytic activity and good recyclability in biomass transformations when compared with various conventional homogeneous systems.
:1 by Zhou's group, and they successfully synthesized the functionalized IL Cr([PSMIM]HSO4)3 and got a perfect yield of HMF.17 Recent studies have demonstrated that the use of homogeneous media such as ionic liquids, may be beneficial for the catalytic conversion of cellulose. Compared to homogeneous systems, heterogeneous systems are easier to separate and recycle, and they have shown superior behavior in terms of yields of HMF. Liu et al. successfully prepared a novel and efficient mesoporous polydivinylbenzene (PDVB) based solid strong acid (PDVB–SO3H–SO2CF3) as a catalyst for biomass transformation.18 Bootsma used propylsulfonic acid and arenesulfonic acid functionalized silicas as organic–inorganic hybrid mesoporous catalysts for cellulose hydrolysis and these were found to catalyze the hydrolysis of cellobiose efficiently, with apparent activation energies similar to those reported previously for homogeneous organic acids.19 The solid catalysts presented excellent catalytic activity and good recyclability in biomass transformations when compared with various conventional homogeneous systems.
Recently, natural clay-particles, which are widely distributed around the world in rock and soil and which possess large specific surface areas, have been considered a potential replacement for macroporous silicon as the support for catalysts in many studies. Dipak adopted clay as a support for heteropoly acids (HPAs) in order to synthesise a composite catalyst and the as prepared composites were successfully applied to the esterification of acetic acid with butanol.20 The clay-based heterogeneous catalysts have many practical and potential applications in green catalysis.21 However, to the best of our knowledge, there are few reports on the use of natural clay-particles in cellulose conversion. Attapulgite (ATP), which possesses an ideal molecular formula of [(Mg,Al)4(Si)8(O,OH,H2O)26·nH2O]22,23 and of which China has large reserves, is a hydrated octahedral layered magnesium aluminum silicate mineral with a fibrous morphology and low cost .24–26 Halloysite nanotubes (HNTs) are an aluminosilicate clay mined from natural deposits and they have a hollow microtubular structure rather than a stacked plate-like structure.27 The well-defined hollow tubular structure of HNTs will be more favorable for the diffusion of cellulose and the acceleration of the catalyst degrading it. Morever, HNTs have a complicated pore structure (20–30 nm), high surface area, and a few hydroxyl groups,28 indicating that some large molecules are easily adsorbed onto their surface.
Up till now, the mechanism of cellulose conversion could be considered to have three main reactions: (1) cellulose-to-glucose depolymerization, (2) glucose-to-fructose isomerization, and (3) fructose-to-HMF dehydration.29 It has been believed over the last few years that acid catalysts are helpful for accelerating the hydrolysis of cellulose, and various acid catalysts have been reported to transform carbohydrates into HMF, such as mineral acids,30 ion exchange resins,31 zeolites32 and metal ions.33
Inspired by the information mentioned above, we report here the successful preparation of acid–chromic chloride functionalized natural clay-particles. Firstly, 3-(mercaptopropyl)trimethoxysilane (MPTMS) was adopted to make the –SH groups to be grafted onto the surface of the ATP and HNTs, respectively. Secondly, 30% H2O2 was used to initiate the oxidation reaction of –SH to –SO3H. After these two steps, the ATP–SO3H and HNTs–SO3H catalysts were prepared. To make the bifunctionalized catalysts, the as prepared ATP–SO3H and HNTs–SO3H were immersed in a solution of CrCl3 to obtain ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III). The morphology and various properties of the synthesized ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) were characterized using SEM, TEM, BET analysis and an Energy Dispersive Spectrometer (EDS). X-ray photoelectron spectroscopy (XPS) measurements were carried out to detect active sites of the synthesized ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III). The acidic features of the ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) solid catalysts were determined by means of Temperature Programmed Desorption (NH3–TPD), TG and analysis of N2 sorption isotherms, and its recyclability in cellulose conversion to HMF was also investigated in our work. Moreover, in order to find the optimal conditions for the system containing the ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) catalysts, the reaction time and temperature were discussed in detail.
2. Experimental section
2.1. Chemicals and reagents
ATP was supplied by Aotebang International Co. in China. HNTs was collected from Zhengzhou Jinyangguang Chinaware Co. Ltd., Henan, China. Prior to use, ATP and HNTs were refluxed with HCl and HNO3, respectively. In a typical procedure 1.0 g of ATP was treated with 100 mL of 8.0 M HCl at 80 °C for 2.0 h, and then cooled to room temperature. The suspension was filtered and thoroughly washed with deionized water to remove Cl−, and then dried at 80 °C for 12 h to form the acid-leached sample (white powder).34 The HNTs (40 g) were treated with 250 mL 3.0 M HNO3 at 75 °C for 12 h, and then cooled to room temperature. After filtration to achieve a pH of 7.0, the obtained powders were calcined at 200 °C for 2.0 h. 1-Ethyl-3-methyl-imidazolium chloride ([EMIM]-Cl), cellulose (powder, ca. 50 micron), 5-HMF (>99%), and 3-(mercaptopropyl)trimethoxysilane (MPTMS) were purchased from Shanghai Aladdin Industrial Inc. Toluene, methanol, ethanol, CrCl3·6H2O, HPLC-grade methanol were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). HCl and 30% H2O2 were obtained from Yangzhou-Shanghai Bao Chemical Co., Ltd. (Shanghai). All other chemicals were supplied by local suppliers and used without further purification.2.2. Functionalization of the ATP and HNTs with acid groups
Synthesis of ATP–SO3H and HNTs–SO3H was implemented by the following steps. Firstly, activated ATP and HNTs were functionalized with hydrosulphide in the presence of MPTMS. Typically, toluene (40 mL) was injected into a three-neck round-bottom flask containing 1.0 g of activated ATP or HNTs, followed by injection of MPTMS (8.0 mL). Then, the mixture was heated and refluxed at 110 °C for 24 h. Finally, the functionalized ATP or HNTs was collected by filtration, washed with toluene several times in order to remove the residual reactant, and dried in vacuum. The resulting samples were called ATP–SH and HNTs–SH. Secondly, ATP–SH and HNTs–SH were oxidized to ATP–SO3H and HNTs–SO3H, respectively.29 Then, 0.5 g of ATP–SH or HNTs–SH was added into a mixture of hydrogen peroxide (10 mL), deionized water (10 mL), and methanol (10 mL). The mixture was stirred at room temperature for 12 h. After that, the resulting precipitate was collected by filtration, washed with deionized water several times and dried under vacuum. The obtained samples resulting from ATP–SH and HNTs–SH were named ATP–SO3H and HNTs–SO3H.2.3. Synthesis of bifunctional catalysts
0.2 g of ATP–SO3H or HNTs–SO3H was added into 10 mL of 0.1 M CrCl3 aqueous solution and then the mixture system was precipitated for 12 hours at room temperature after ultrasonication. Finally, the bifunctional catalysts ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) were collected by filtration, washed with deionized water several times and dried in vacuum in order to obtain powders.2.4. Characterization of ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III)
The morphologies of ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) were observed using a transmission electron microscope (TEM, JEOL IEM-200CX) and scanning electron microscope (SEM, S-4800). TGA of the samples were performed for powder samples (about 10 mg) using a Diamond TG/DTA instrument (Perkin-Elmer, U.S.A.) under a nitrogen atmosphere up to 800 °C with a heating rate of 5.0 °C min−1. XPS was performed using a Thermo ESCALAB 250 with Al Kα radition at y = 901 for the X-ray sources, and the binding energies were calibrated using the C1s peak at 284.9 eV. The acidic features of the ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) solid catalysts were determined by means of temperature programmed desorption (NH3–TPD), using a chromatographic thermal conductivity detector, NH3 as the adsorbate, and He as the carrier gas. For a typical run, 0.2 g of catalyst (40–60 mesh) was saturated with NH3 at 30 °C for 45 min. Then, the sample was exposed to the flowing He in order to remove physically adsorbed ammonia on the surface of the sample. Finally, desorption of NH3 was carried out by heating the sample from 30 °C to 700 °C. Desorption of NH3 was analyzed by gas chromatography equipped with a thermal conductivity detector. NH3 adsorption–temperature curves were plotted, and by comparing the stripping peak temperatures we obtained the strength of the acid center of the sample. We can also get the intensity of different acid samples by analyzing the peak area of the center.2.5. Typical procedure for cellulose conversion
Experiments were carried out in a 25 mL graduated pyrex glass tube immersed in an oil bath preheated at the required temperature using a hot plate with a digital magnetic stirrer. The cellulosic conversion included two steps of pre-treatment and reaction. In a typical experiment for pre-treatment, cellulose (0.1 g) was added into [EMIM]-Cl (2.0 g), and the whole mixture was heated at 120 °C and stirred at 800 rpm for 0.5 h for the dissolution of cellulose. At the reaction step, catalyst (0.1 g) was added into the cellulose–[EMIM]Cl solution whilst heating and stirring were maintained at 120 °C and 800 rpm, respectively, for another 2.0 h. All reaction steps were repeated three times. Finally, the concentration of HMF was analyzed by HPLC, and the average yields of products were calculated.2.6. HMF analysis
The HMF was analyzed using a 1200 Agilent High Performance Liquid Chromatography (HPLC) system equipped with an Agilent TC-C18(2) column (4.6 × 250 mm, 5.0 μm) and UV detector. During this process, the column temperature remained constant at 30 °C, and the mobile phase was methanol–water (7:3, v/v) with a flow rate of 0.7 mL min−1 with UV detection at 283 nm, and 22.5 μL of each sample was also injected manually. The concentration of HMF was calculated based on the standard calibration curve obtained with standard prepared solutions containing HMF.
3. Results and discussion
3.1. Characterization of ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III)
The morphology and various properties of the synthesized ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) were characterized using SEM, TEM, an Energy Dispersive Spectrometer (EDS) and nitrogen adsorption–desorption. As shown in Fig. 1a, ATP–SO3H–Cr(III) emerged in the form of fibers with an average diameter of about 40–60 nm, while the HNTs–SO3H–Cr(III) exhibited a tubular morphology with an average diameter of about 80–100 nm, as shown in the Fig. 1b. From Fig. 1c, it could be considered that HNTs–SO3H–Cr(III) had an open-ended structure. Fig. 1d and e represent the EDS analysis of ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III), respectively. According to the results shown in Fig. 1d and e, both ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) possessed the peaks for elements S and Cr, indicating the presence of the sulfonic acid group and Cr(III) in the two catalysts. After calculating the weights of the elements present, S was found to be 1.60% and 3.99%, and Cr was 1.48% and 2.21% in the ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III), respectively. Moreover, the peak of element Al could be assigned to the raw materials of ATP and HNTs.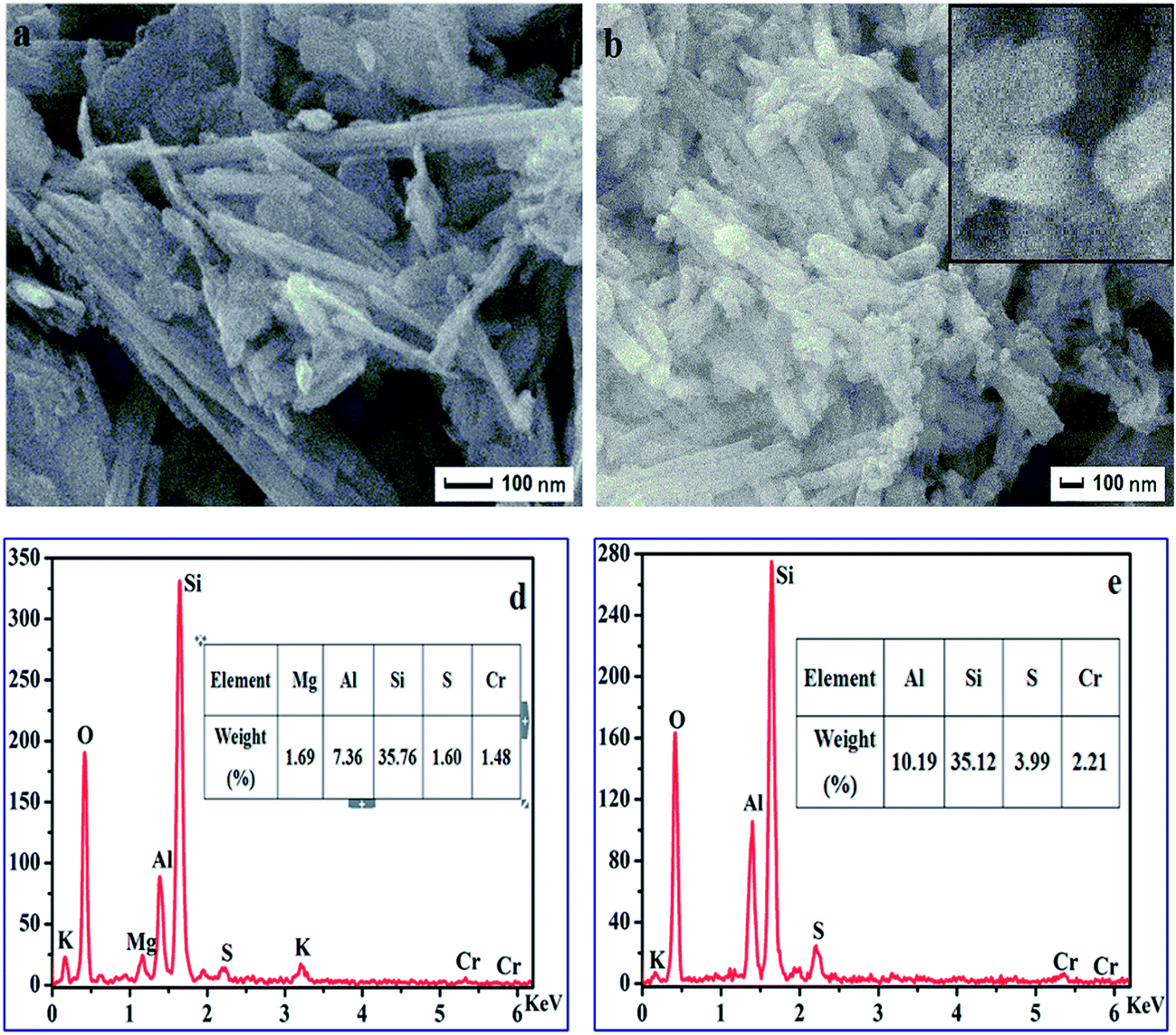 | ||
| Fig. 1 SEM of ATP–SO3H–Cr(III) (a) and HNTs–SO3H–Cr(III) (b and c). Energy Dispersive Spectrometer (EDS) spectra of ATP–SO3H–Cr(III) (d) and HNTs–SO3H–Cr(III) (e). | ||
To further study the morphology and surface properties, TEM images of ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) are shown in Fig. 2a and b, and their close-up images are also shown in Fig. 2b and d, respectively. In Fig. 2a there are some detectable holes on the surface of ATP–SO3H–Cr(III), which could allow the targets to go through the surface and reach the active sites. It can also be seen that HNTs–SO3H–Cr(III) has an open-ended structure, and the nanotubes have an average width in the range 65–100 nm and an inner diameter of about 30 nm. Moreover, a few grafted –SO3H groups and Cr(III) were adhered to the external surface of HNTs, and the hollow tubes were also filled with grafted –SO3H groups and Cr(III), which is beneficial for improving the active sites.
 | ||
| Fig. 2 TEM of ATP–SO3H–Cr(III) (a and b) and HNTs–SO3H–Cr(III) (c and d). | ||
Fig. 3 shows the nitrogen adsorption–desorption isotherms of ATP (Fig. 3a), ATP–SO3H–Cr(III) (Fig. 3b), HNTs (Fig. 3c), and HNTs–SO3H–Cr(III) (Fig. 3d). Clearly, all of the samples exhibit typical type-IV isotherms, giving a steep increase at a relative pressure between 0.8 < P/P0 < 1.0. Additionally, the nitrogen adsorption–desorption isotherms also showed that the BET surface area of ATP was 104.46 m2 g−1 and HNTs was 151.58 m2 g−1, while ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) give BET surface areas of 78.29 m2 g−1 and 63.85 m2 g−1, respectively, indicating that more groups have been grafted onto the HNTs than ATP.
 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms of (a) ATP, (b) ATP–SO3H–Cr(III), (c) HNTs and (d) HNTs–SO3H–Cr(III). | ||
3.2. Active site characterizations
Fig. 4 shows the X-ray photoelectron spectroscopy (XPS) measurements for ATP (Fig. 4a), ATP–SO3H–Cr(III) (Fig. 4b), HNTs (Fig. 4e) and HNTs–SO3H–Cr(III) (Fig. 4f). Fig. 4c, d, g and h show the magnified signals of S2p and Cr2p3. Clearly, both ATP and ATP–SO3H–Cr(III) exhibited the signals for Mg and Si, while both the HNTs and HNTs–SO3H–Cr(III) showed the signals for Al and Si, indicating the presence of ATP and HNTs in ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III), respectively. Compared to ATP, two new signals of S2p and Cr2p3 can also be observed in ATP–SO3H–Cr(III), confirming the successful grafting of –SO3H and Cr(III) onto the raw ATP particles. The same phenomenon was also observed by carrying out a comparative study of HNTs and HNTs–SO3H–Cr(III). According to the previous work, the existence of the –SO3H group can provide the abundant acidic sites, and the introduction of Cr(III) can enhance the catalytic performance in the conversion of cellulose to HMF.35 Thus, an acid–chromic chloride functionalized natural clay may be an effective catalyst for the degradation of cellulose to HMF. | ||
| Fig. 4 X-ray photoelectron spectroscopy (XPS) of ATP (a), ATP–SO3H–Cr(III) (b), HNTs (e), and HNTs–SO3H–Cr(III) (f). | ||
3.3. Acidic features of ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III)
The acidic features of obtained solid catalysts were determined by means of temperature programmed desorption (NH3–TPD). The NH3–TPD curves of ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) are shown in Fig. 5a and b, respectively. The acid sites based on desorption temperature were divided into three categories: weak acid (150 °C–250 °C), medium acid (250 °C–350 °C), and strong acid (350 °C–450 °C).36 Interestingly, ATP–SO3H–Cr(III) exhibited two sharp NH3 desorption peaks centered at 250 °C and 450 °C, indicating the presence of moderate acidic sites and strong acidic sites, respectively. Compared to ATP–SO3H–Cr(III), it was clearly seen that HNTs–SO3H–Cr(III) had three peaks around 450 °C, 550 °C, and 600 °C, which could be assigned to the strong acidic sites. As a result, HNTs–SO3H–Cr(III) possessed a much stronger acid strength and more strong acidic sites. | ||
| Fig. 5 Temperature programmed desorption (NH3–TPD) of ATP–SO3H–Cr(III) (a) and HNTs–SO3H–Cr(III) (b). | ||
TG analysis was employed to estimate the weight ratio of inorganic and organic components, as shown in Fig. 6a for ATP–SO3H–Cr(III) and Fig. 6b for HNTs–SO3H–Cr(III). It could be clearly seen that both of the catalysts exhibited weight loss between the temperature ranges 25 °C–200 °C and 200 °C–800 °C, which were associated with the loss of H2O contained in the catalysts and the decomposition of functional groups i.e. –SO3H, respectively.37 Notably, the weight loss of the organic components between 200 °C and 800 °C was 8.07% for ATP–SO3H–Cr(III) and 13.77% for HNTs–SO3H–Cr(III), indicating the successful grafting of the –SO3H group onto the surface of ATP and HNTs. What is more, the greater loss of organic components from ATP–SO3H–Cr(III) than from HNTs–SO3H–Cr(III) could also be explained by the fact that the amount of –SO3H grafted onto the HNTs was higher than that of ATP. This is consistent with the results of the EDS analysis which showed a greater amount of S in HNTs–SO3H–Cr(III) than in ATP–SO3H–Cr(III). This phenomenon may result in the higher yield of HMF for HNTs–SO3H–Cr(III) than that for ATP–SO3H–Cr(III).
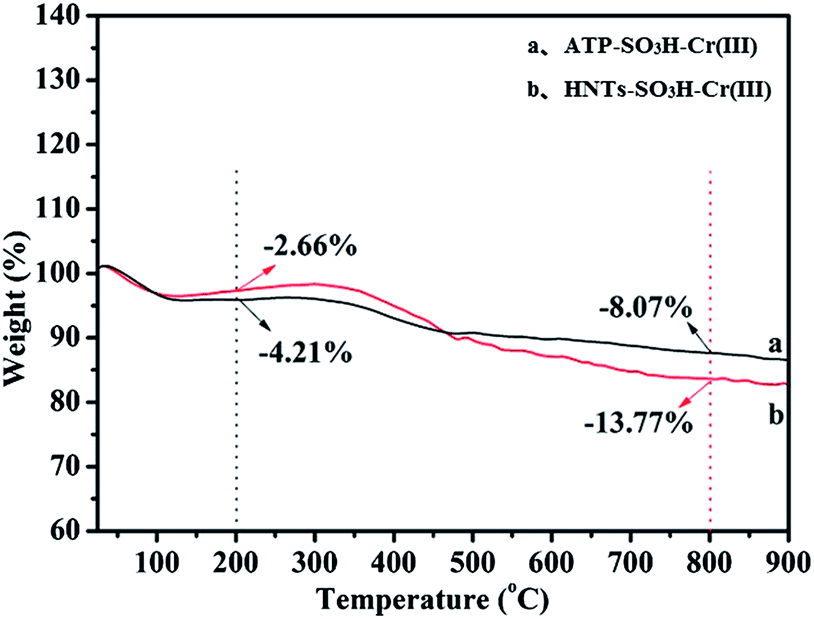 | ||
| Fig. 6 TG curves of (a) ATP–SO3H–Cr(III) and (b) HNTs–SO3H–Cr(III). | ||
3.4. Catalytic activities
Cellulosic conversion involves a series of catalytic reactions, including (1) cellulose-to-glucose depolymerization, (2) glucose-to-fructose isomerization, and (3) fructose-to-HMF dehydration.29 In order to find the optimal conditions for the system containing ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) catalysts, the reaction time and temperature were considered the most important conditions in this work.17,38,39So far, a large amount of literature has reported that a reaction time of 2.0 h is very beneficial to the degradation of cellulose.40–42 Though the complete degradation of cellulose needs longer than the optimal amount of time, the HMF yield decreases when the reaction time is above 2.0 h. Firstly, we took a relatively low temperature (≦130 °C), as small molecules (glucose, cellobiose, HMF, furfural et al.) can be easily obtained from cellulose depolymerization in [BMIM]-Cl at this temperature.17 Therefore, the most suitable temperature for this reaction with these catalysts is 120 °C–130 °C. Thus, we took the reaction time of 2.0 h and temperatures of 100 °C, 120 °C and 140 °C. We obtained HMF yields of 15.30% and 18.72% at 100 °C for ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III), respectively, and 22.27% and 28.13% at 140 °C for ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III), respectively, both sets of values are lower than those achieved at 120 °C. We can draw the conclusion that a temperature of 120 °C was beneficial for cellulose degradation catalyzed by ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III). It is suggested that increasing the reaction temperature can decrease the viscosity of the reaction system, and accelerate the mass transfer rate, which is in favor of promoting the reaction rate and the dehydration of cellulose to HMF. However, as the temperature is increased, some side reactions prefer to occur when the temperature is close to the melting point of monosaccharides, such as the formation of insoluble humines and soluble polymers (decomposition products of fructose).43–45 In Tao's work, which involved catalyzing the cellulose with SO3H-functionalized ionic liquids (0.5 g) for 300 min at 150 °C, the conversion of microcrystalline cellulose (MCC) reached up to 78.5%, and the yields of HMF and furfural were up to 29.7% and 16.9%, respectively, using longer reaction times and higher reaction temperatures but obtaining a lower yield of HMF than that of our work.46
After finding out the optimal reaction temperature, we discussed another important factor and investigated the reaction time ranging from 1.0 h to 5.0 h keeping the temperature constant at 120 °C. Two systems contained the same dose of cellulose (0.1 g) and ILs (2.0 g) but different catalysts of ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III). As shown in Fig. 7, it could be found that the yield increased with increasing reaction time at first. After reaching the time of 2.0 h, the yields from all of the catalysts decreased when the reactions were continued. It was suggested that this was due to the decomposition of HMF to other byproducts. Some possible byproducts such as formic acid and levulinic acid were examined by us, but could not be detected. Several papers have also reported the decomposition of HMF to some unidentified products.43–45 Thus, the optimal reaction conditions for both ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) are a temperature of 120 °C and a reaction time of 2.0 h, with the highest yields of HMF at 31.20% and 41.22%, respectively. We can clearly see that HNTs–SO3H–Cr(III) has a better catalytic performance for cellulose conversion than ATP–SO3H–Cr(III). This kind of phenomenon thoroughly supports the situation observed in Fig. 1d and e, which show that the HNTs–SO3H–Cr(III) catalyst contained larger amounts of the –SO3H group and Cr(III) than the ATP–SO3H–Cr(III) catalyst. What is more, we also used ATP–SO3H and HNTs–SO3H as catalysts to degrade the cellulose to HMF under the optimal reaction conditions which we ascertained. Just as we anticipated, we obtained yields of HMF for ATP–SO3H of 24.79% and HNTs–SO3H of 26.68%. This result indicates that the –SO3H group and Cr(III) have co-catalytic performance when used as catalysts for cellulose degradation.
 | ||
| Fig. 7 Effect of time on the yield of cellulose to HMF. | ||
3.5. Recyclability of the catalysts
To study the recyclability of the synthesized bi-functional materials, ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) were selected to study their reusability for the cellulose dehydration reaction over five cycles. The two catalysts were recovered both by centrifugation after immersion with 0.1 M of HCl for 12 hours. After filtration, the residue was dried at 80 °C to obtain the catalysts as powders. All reaction conditions were the same as the optimized conversion conditions. As shown in Fig. 8a, the yields of HMF remained at around 21.50% and 22.35% until the fifth run for ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III), respectively. Additionally, no significant decrease in the HMF yield was observed till the third run for ATP–SO3H–Cr(III) and the second run for HNTs–SO3H–Cr(III). This means that the immobilized functional groups of –SO3H and Cr(III) did not leach significantly during the complicated and repeated process. What is more, the TEM and EDS analyses of the catalysts used in the third run are shown in Fig. 8b and c and represent the morphology of ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) after being used three times. From the images, it can be seen that the structures of both ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) are not damaged. In comparison with Fig. 1d and e, in Fig. 8d and e it could be observed that the amount of Cr(III) has decreased greatly while the amount of S has reduced slightly. The diminishing yield of HMF can be attributed to the decrease in the amount of Cr(III). These results, to some extent, suggest that the heterogeneous catalysts have the potential to be effectively separated and reused for the dehydration reaction. | ||
| Fig. 8 (A) Recyclability of ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) in the cellulose-to-HMF conversion. Reaction conditions: cellulose = 0.1 g, IL = 2.0 g, catalyst = 0.1 g, T = 120 °C, t (pre-treatment) = 0.5 h, t (reaction) = 2 h (a). (B) SEM showing the morphology of ATP–SO3H–Cr(III) (b) and HNTs–SO3H–Cr(III) (c) after being used three times. (C) Energy Dispersive Spectrometer (EDS) image of ATP–SO3H–Cr(III) (d) and HNTs–SO3H–Cr(III) (e) after being used three times. | ||
4. Conclusions
In this work, we firstly demonstrated the synthesis of ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) and then discussed their characterization using SEM, TEM, EDS, XPS, BET, NH3–TPD and TG analysis. These two catalysts showed enhanced catalytic efficacy toward cellulose-to-HMF conversion, HNTs–SO3H–Cr(III), especially, can achieve a yield of 41.22% under the optimal conditions. Using clay as a support for the catalytic reaction can also lower the price of the catalytic process. In addition, the prepared catalysts can be very easily recycled at least three times without significant loss of activity. We expect that the bi-functionalized and recyclable ATP–SO3H–Cr(III) and HNTs–SO3H–Cr(III) catalysts could also be useful for other catalytic applications.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21107037, no. 21176107, no. 21306013, no. 21306013), Natural Science Foundation of Jiangsu Province (no. BK2011461, no. BK2011514), National Postdoctoral Science Foundation (no. 2013M530240), Postdoctoral Science Foundation funded Project of Jiangsu Province (no. 1202002B) and Programs of Senior Talent Foundation of Jiangsu University (no. 12JDG090).References
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS PubMed.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- Y. Sun and J. Y. Cheng, Bioresour. Technol., 2002, 83, 1–11 CrossRef CAS.
- M. Stocker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef CAS PubMed.
- C. W. Hu, X. Xiang, L. He, Y. Yang, B. Guo and D. M. Tong, Catal. Lett., 2011, 141, 735–741 CrossRef.
- O. O. James, S. Maity, L. A. Usman, K. O. Ajanaku, O. O. Ajani, T. O. Siyanbola, S. Sahu and R. Chaubey, Energy Environ. Sci., 2010, 3, 1833–1850 CAS.
- M. Balakrishnan, E. R. Sacia and A. T. Bell, Green Chem., 2012, 49, 1626–1634 RSC.
- S. M. Notley, B. Pettersson and L. Wagberg, J. Am. Chem. Soc., 2004, 126, 13930–13931 CrossRef CAS PubMed.
- I.-J. Kuo, N. Suzuki, Y. Yamauchi and K. C.-W. Wu, RSC Adv., 2013, 3, 2028–2034 RSC.
- C. Li and Z. K. Zhao, Adv. Synth. Catal., 2007, 349, 1847–1850 CrossRef CAS.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- Z. Zhang and Z. K. Zhao, Bioresour. Technol., 2010, 101, 1111–1114 CrossRef CAS PubMed.
- F. Jiang, Q. J. Zhu, D. Ma, X. M. Liu and X. W. Han, J. Mol. Catal. A: Chem., 2011, 334, 8–12 CrossRef CAS PubMed.
- K. Seri, T. Sakaki, M. Shibata, Y. Inoue and H. Ishida, Bioresour. Technol., 2002, 81, 257–260 CrossRef CAS.
- A.-Y. Hussein, E. B. Hassan and P. Steele, J. Fuel Chem. Technol., 2013, 41, 214–222 CrossRef.
- L. L. Zhou, R. J. Liang, Z. W. Ma, T. H. Wu and Y. Wu, Bioresour. Technol., 2013, 129, 450–455 CrossRef CAS PubMed.
- F. J. Liu, A. M. Zheng, I. Noshadi and F. S. Xiao, Appl. Catal., B, 2013, 136–137, 193–201 CrossRef CAS PubMed.
- J. A. Bootsma and B. H. Shanks, Appl. Catal., A, 2007, 327, 44–51 CrossRef CAS PubMed.
- S. K. Bhorodwaj and D. K. Dutta, Appl. Clay Sci., 2011, 53, 347–352 CrossRef CAS PubMed.
- C. H. Zhou, Appl. Clay Sci., 2011, 53, 87–96 CrossRef CAS PubMed.
- R. Giustetto, F. X. L. Xamena, G. Ricchiardi, S. Bordiga, A. Damin, R. Gobetto and M. R. Chierotti, J. Phys. Chem. B, 2005, 109, 19360–19368 CrossRef CAS PubMed.
- J. Huang, Y. Liu, Q. Jin, X. Wang and J. Yang, J. Hazard. Mater., 2007, 143, 541–548 CrossRef CAS PubMed.
- L. M. Shi, J. F. Yao, J. L. Jiang, L. X. Zhang and N. P. Xu, Microporous Mesoporous Mater., 2009, 122, 294–300 CrossRef CAS PubMed.
- J. M. Pan, X. H. Zou, X. Wang, W. Guan, Y. S. Yan and J. Han, Chem. Eng. J., 2010, 162, 910–918 CrossRef CAS PubMed.
- J. M. Pan, X. H. Zou, Y. S. Yan, X. Wang, W. Guan, J. Han and X. Y. Wu, Appl. Clay Sci., 2010, 50, 260–265 CrossRef CAS PubMed.
- S. R. Levis and P. B. Deasy, Int. J. Pharm., 2002, 243, 125–134 CrossRef CAS.
- X. M. Sun, Y. Zhang, H. B. Shen and N. Q. Jia, Electrochim. Acta, 2010, 56, 700–705 CrossRef CAS PubMed.
- W. H. Peng, Y. Y. Lee, C. Wu and K. C.-W. Wu, J. Mater. Chem., 2012, 22, 23181–23185 RSC.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2007, 46, 7703–7710 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Ind. Eng. Chem. Res., 2008, 47, 9234–9239 CrossRef CAS.
- C. Moreau, R. Durand, S. Razigade, J. Duhamet, P. Faugeras, P. Rivalier, P. Ros and G. Avignon, Appl. Catal., A, 1996, 145, 211–224 CrossRef CAS.
- H. B. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- H. M. Yang, A. D. Tang, J. Ouyang, M. Li and S. Mann, J. Phys. Chem. B, 2010, 114, 2391–2396 Search PubMed.
- H. Li, Q. Y. Zhang, X. F. Liu, F. Chang, Y. P. Zhang, W. Xue and S. Yang, Bioresour. Technol., 2013, 144, 21–27 CrossRef CAS PubMed.
- G. D. Yadav and A. D. Murkute, Adv. Synth. Catal., 2004, 346, 389–394 CrossRef CAS.
- F. J. Liu, X.-J. Meng, Y. L. Zhang, L. M. Ren, F. Nawaz and F.-S. Xiao, J. Catal., 2010, 271, 52–58 CrossRef CAS PubMed.
- H. Li, Q. Y. Zhang, X. F. Liu, F. Chang, D. Y. Hu, Y. P. Zhang, W. Xue and S. Yang, RSC Adv., 2013, 3, 3648–3654 RSC.
- Y. Yang, M. M. Abu-Omar and C. W. Hua, Appl. Energy, 2012, 99, 80–84 CrossRef CAS PubMed.
- Y. Su, H. M. Brown, X. W. Huang, X. D. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117–122 CrossRef CAS PubMed.
- W. H. Hsu, Y. Y. Lee, W. H. Peng and K. C.-W. Wu, Catal. Today, 2011, 174, 65–69 CrossRef CAS PubMed.
- M. X. Tan, L. Zhao and Y. G. Zhang, Biomass Bioenergy, 2011, 35, 1367–1370 CrossRef CAS PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- A. I. Torres, P. Daoutidis and M. Tsapatsis, Energy Environ. Sci., 2010, 3, 1560–1572 CAS.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- F. R. Tao, H. L. Song and L. J. Chou, Bioresour. Technol., 2011, 102, 9000–9006 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |