An efficient and green procedure for synthesis of rhodanine derivatives by aldol-thia-Michael protocol using aqueous diethylamine medium†
Assem Barakat*ab,
Abdullah M. Al-Majida,
Hany J. AL-Najjara,
Yahia N. Mabkhota,
Hazem A. Ghabbourc and
Hoong-Kun Func
aDepartment of Chemistry, College of Science, King Saud University, P. O. Box 2455, Riyadh 11451, Saudi Arabia. E-mail: ambarakat@ksu.edu.sa; Fax: +966 1-4675992; Tel: +966 1-4675884
bDepartment of Chemistry, Faculty of Science, Alexandria University, P.O. Box 426, Ibrahimia, 21321 Alexandria, Egypt
cDepartment of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, P. O. Box 2457, Riyadh 11451, Saudi Arabia
First published on 9th December 2013
Abstract
A simple, economical, and green approach to the synthesis of rhodanine derivatives using a tandem aldol condensation-thia-Michael addition process in aqueous diethylamine medium was described. The experiment protocol features simple operations, and the products were isolated in high to excellent yields (82–96%). As spontaneous precipitation always occurs at the end of the process, this leads to easy separation of the products via a simple filtration.
Introduction
The five-membered rhodanine core is an interesting heterocyclic ring system featured in a large number of natural or synthetic compounds with a wide range of pharmacological activities.1 Rhodanine derivatives have been reported as small molecule inhibitors for targets such as hepatitis C virus NS5B polymerase2 and human cathepsin D.3 Furthermore, they have been reported as antimalarial, antiviral, antibacterial, and anti-cancer, and antidiabetic agents (Fig. 1).4 Epalrestat is a well-known and highly marketed drug comprising the rhodanine nuclei and used to delay the progression of diabetic neuropathy.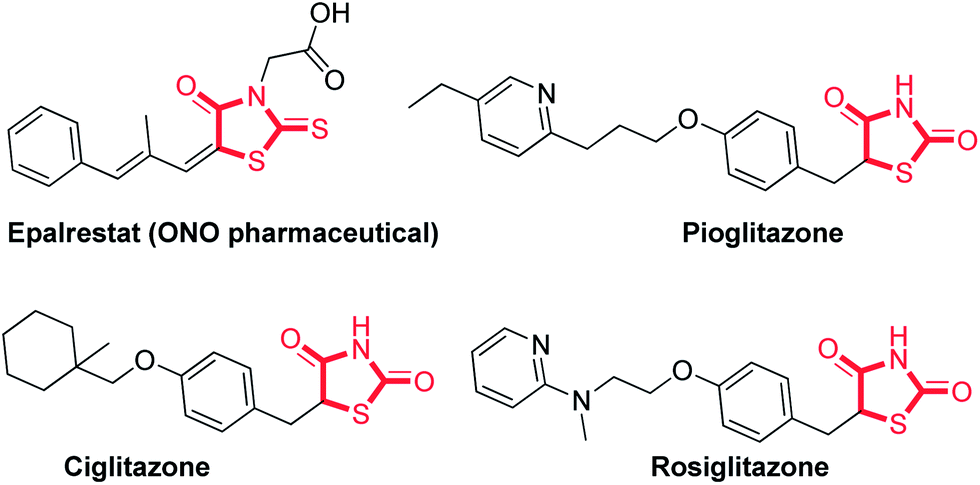 | ||
| Fig. 1 Bioactive compounds containing the rhodanine and thiazolidinedione framework.5 | ||
It is therefore not surprising that numerous synthetic routes have been developed to obtain this heterocyclic core, many of which were recently reported.6 Due to stringent and growing environmental regulations, organic chemists have endeavored to develop clean, economical, and environmentally safer methodologies.7 Current emphasis on the development of multiple chemical transformations sequentially performed in a single reaction vessel, without intermediary purification steps, has led to the generation of a variety of molecular complexity. The benefits of the green approach are based on reduced time, costs, and waste generation, but compatibility and reliability must be circumvented for such processes to become attractive for industrial purposes. During the past decade, aldol-thia-Michael protocol is an important process in organic chemistry and has versatile applications in organic synthesis.8 Different catalysts and reaction media have been employed, such as Lewis acids, cinchona alkaloids, ionic liquids, and solid support.9 One of the most promising approaches is using water as the reaction medium as reported by Saeed Abaee, M. et al. on a multicomponent synthesis of β-aryl-β-mercapto ketones.10
With this in mind and in continuation of our research program on the developments of new routes to heterocyclic system,11 we now report the aldol-thia-Michael addition process for the synthesis of rhodanine derivatives where water is used as a green solvent in the presence of diethylamine. These products were subjected for biological and pharmacological evaluation.
Results and discussion
In a preliminary experiment, treatment of 2-thioxothiazolidin-4-one (1) with benzaldehyde (2) using water–diethylamine at room temperature for 3 h (TLC control) afforded the (Z)-5-benzylidene-2-thioxothiazolidin-4-one adduct (3a·HNEt2).12 The configuration of its double bond was determined on the basis of X-ray diffraction analysis of its salt with diethylamine (Fig. 2). Subsequently, addition of thiophenol to the mixture at this point led to quantitative consumption of the reactants within a few minutes and the formation of 4a·HNEt2 in 96% yield (entry 1). The structures of products 3a·HNEt2 and 4a·HNEt2 were identified by their spectroscopy analysis. The structure of 4a·HNEt2 was further confirmed by X-ray diffraction analysis. The molecular structure of 4a·HNEt2 is shown in Fig. 3. Interestingly, the X-ray shown that 4a·HNEt2 has been formed with a molecule of diethylamine as a salt (Fig. 3). Encouraged by this result, we proceeded to study the effect of different amines and reaction conditions on the aldol-thia-Michael addition process for the synthesis of rhodanines derivatives.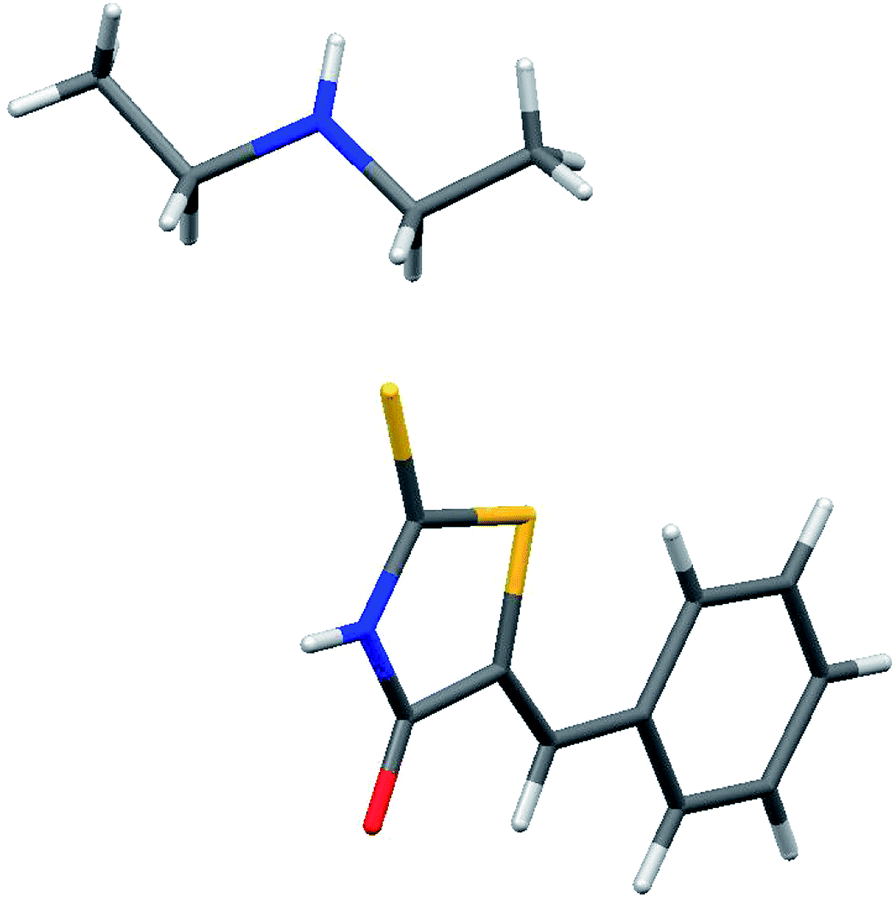 | ||
| Fig. 2 X-ray diffraction structure of compound 3a·HNEt2. | ||
 | ||
| Fig. 3 X-ray diffraction structure of compound 4a·HNEt2. | ||
As shown in Table 1, it was found that iPr2NH–H2O afford the aldol-thia-Michael adduct in excellent yield 89% (Table 1, entry 2). Other secondary amines, such as (cyclohexyl)2NH and morpholine, behaved similarly and generated the respective products (Table 1, entry 3 and 4) but showed lower efficiencies as evidenced by the 83% and 80% yield respectively. NaOH was also tested, and was found to be less efficient in the reaction; only moderate yield was obtained (Table 1, entries 5). The products could not be obtained in the absence of either amine (entry 6) or water (entry 7), i.e. the reaction either could not be processed or preceded too slowly to be detected. Collectively, the best result with respect to yield was obtained by performing the reaction by the combined promoting effects of both water and diethylamine (Scheme 1).
| Entry | Condition | Time [h] | Yieldb (%) |
|---|---|---|---|
| a All reactions were carried out with 2-thioxothiazolidin-4-one 1 (3.0 mmol), benzaldehyde 2 (3.0 mmol) and diethylamine (3.0 mmol) in water (2–3 ml) for the specified time for aldol condensation.b Yield of isolated product 4a·HNEt2. | |||
| 1 | Et2NH–H2O | 3 | 96 |
| 2 | iPr2NH–H2O | 4 | 89 |
| 3 | (Cyclohexyl)2NH–H2O | 4 | 83 |
| 4 | Morpholine–H2O | 3 | 80 |
| 5 | NaOH–H2O | 6 | 70 |
| 6 | Et2NH | 10 | 10 |
| 7 | H2O | 10 | 0 |
 | ||
| Scheme 1 Model substrate during the optimization studies. | ||
A plausible mechanism can be proposed for the reaction as shown in Fig. 4. First, the hydrogen bonding activation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group by water eases up the deprotonation of 1 by diethylamine. The aldehyde is then attacked by the enolate to form the aldol condensation product after dehydration of the intermediate. The second thia-Michael addition under these conditions produces the final product 4 (Fig. 4).13
O group by water eases up the deprotonation of 1 by diethylamine. The aldehyde is then attacked by the enolate to form the aldol condensation product after dehydration of the intermediate. The second thia-Michael addition under these conditions produces the final product 4 (Fig. 4).13
 | ||
| Fig. 4 A possible mechanistic pathway. | ||
To support the proposed mechanism, the reaction was stopped before the addition of the thiophenol and after TLC showed complete consumption of aldehyde and 2-thioxothiazolidin-4-one. Analysis of the reaction mixture showed the presence of the single product 3·HNEt2 in quantitative yield. Furthermore, the intermediate products 3b,c·HNEt2 were separated and confirmed by X-ray crystal structure determination (see ESI).†
In continuation of our on going program aimed at the synthesis of rhodanine derivatives, the substrate scope was then investigated. We applied the conditions to reactions of 1 with a variety of other aldehydes followed by addition thiophenol. As revealed in Table 2, (entry 1–18). The reactions proved to work well with a range of aldehydes bearing either electron-withdrawing or electron-donating groups to give the rhodanine derivatives 4 with very good to excellent yields (82–96%). Depending on the structure of the reacting aldehyde the products were isolated as adduct with diethylamine (Table 2, entries 1, 4, 6, 10, 12, 14, and 17) or as individual compounds (entries 2, 3, 5, 7–9, 11, 13, 15, 16 and 18). In the case of adducts 4·HNEt2, the corresponding free rhodanine derivatives 4 can be obtained by treatment with 10% aqueous HCl.
|
|||
|---|---|---|---|
| Entry | Product | R | Yield (%) |
| a All reactions were carried out with 2-thioxothiazolidin-4-one 1 (3.0 mmol), aldehyde 2 (3.0 mmol) and diethylamine (3.0 mmol) in water (2–3 ml) for the specified time.b Yield of the product isolated directly from the reaction mixture (GP1).c Yield of isolated product 4 obtained by treatment of the corresponding salt 4·HNEt2 with aqueous HCl (10%) followed by extraction with DCM/EtOH (GP2). | |||
| 1 | 4a·HNEt2 | Ph | 96b |
| 2 | 4a | Ph | 93c |
| 3 | 4b | p-CH3Ph | 95c |
| 4 | 4c·HNEt2 | p-ClPh | 92b |
| 5 | 4c | p-ClPh | 91c |
| 6 | 4d·HNEt2 | p-BrPh | 90b |
| 7 | 4d | p-BrPh | 90c |
| 8 | 4e | p-CH3OPh | 88c |
| 9 | 4f | 2,4,6-(CH3)3Ph | 83c |
| 10 | 4g·HNEt2 | p-NO2Ph | 85b |
| 11 | 4g | p-NO2Ph | 85c |
| 12 | 4h·HNEt2 | Naphthyl | 91b |
| 13 | 4h | Naphthyl | 90c |
| 14 | 4i·HNEt2 | 2,4-Cl2Ph | 82b |
| 15 | 4i | 2,4-Cl2Ph | 81c |
| 16 | 4j | 2,6-Cl2Ph | 80c |
| 17 | 4k·HNEt2 | m-CH3Ph | 89b |
| 18 | 4k | m-CH3Ph | 87c |

Interesting and surprising was the fact that a variety of heterocyclic aldehydes reacted under the optimized conditions to always yield exclusively the product 3. A possible explanation of such behaviour is that the heterocyclic fragment is probably more conjugated with the double bond because of less steric factor due to smaller cycles (Table 3, entry 1–3) and/or absence of one ortho-proton (Table 3, entry 4). As summarized in Table 3, various heterocyclic aldehyde such as thiophene-2-carbaldehyde, furan-2-carbaldehyde, 1H-pyrrole-2-carbaldehyde and picolinaldehyde were reacted with 2-thioxothiazolidin-4-one using diethylamine–water medium afforded mainly product 5 in excellent yield (91–95%). It has been observed that when furan-2-carbaldehyde was used, the product was a mixture of 3 and 5 (Table 3, entry 2). 1HNMR spectrum shown a characteristic broad singlet at δ 13.67 ppm and δ 8.1 ppm which is assigned to the proton of thiol tautomer 5 and the proton of NH tautomer 3 respectively.




Conclusions
In summary, a general and efficient procedure has been developed for facile synthesis of rhodanine derivatives. Reactions occur under mild aqueous conditions using quantities of diethylamine. More importantly, the products precipitate spontaneously in the mixture allowing their convenient purification without costly and time consuming chromatographic separations. The reactions gave high yields of products in short times. The full scope, asymmetric transformations and its applications in the synthesis of biologically active molecules are currently underway in our laboratory.Experimental section
General: All the chemicals were purchased from Aldrich, Sigma-Aldrich, Fluka etc., and were used without further purification, unless otherwise stated. All melting points were measured on a Gallenkamp melting point apparatus in open glass capillaries and are uncorrected. IR Spectra were measured as KBr pellets on a Nicolet 6700 FT-IR spectrophotometer. The NMR spectra were recorded on a Varian Mercury Jeol-400 NMR spectrometer. 1H-NMR (400 MHz), and 13C-NMR (100 MHz) were run in either deuterated dimethylsulphoxide (DMSO-d6) or deuterated chloroform (CDCl3). Chemical shifts (δ) are referred in terms of ppm and J-coupling constants are given in Hz. Mass spectra were recorded on a Jeol of JMS-600H. Elemental analysis was carried out on Elmer 2400 Elemental Analyzer; CHN mode.General procedure for aldol condensation thia-Michael addition for the synthesis of 4·HNEt2 (GP1)
A mixture of aldehyde 2 (3 mmol, 318 mg), 2-thioxothiazolidin-4-one 1 (3 mmol, 400 mg), and diethylamine (3 mmol, 310 μl) in 3 ml of degassed H2O was stirred at room temperature for 3–5 hours until TLC showed complete disappearance of the reactants. The thiol (3 mmol, 330 mg) was added to this mixture and stirring was continued for another 4–6 min until TLC showed completion of the reaction. The product precipitated and the mixture was filtered and the solid portion was recrystallized from a mixture of DCM/EtOH to obtain the pure product 4·HNEt2.General procedure for aldol condensation thia-Michael addition for the synthesis of 4 (GP2)
A mixture of aldehyde 2 (3 mmol, 318 mg), 2-thioxothiazolidin-4-one 1 (3 mmol, 400 mg), and diethylamine (3 mmol, 310 μl) in 3 ml of degassed H2O was stirred at room temperature for 3–5 hours until TLC showed complete disappearance of the reactants. The thiol (3 mmol, 330 mg) was added to this mixture and stirring was continued for another 4–6 min until TLC showed completion of the reaction. Water (3 ml) was the added into the reaction mixture, then extracted with DCM/EtOH (3 × 50 ml) and the organic phase washed with 10% HCl (2 × 50 ml), followed by brine (2 × 50 ml), and dried over MgSO4, and filtered and evaporated to afford 4.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) (4a·HNEt2). 4a·HNEt2 was prepared from 2-thioxothiazolidin-4-one 1, benzaldehyde and thiophenol according to the general procedure (GP1) yielding yellow crystalline materials (1.17 g, 2.9 mmol, 96%). m.p: 180 °C; IR (KBr, cm−1): 3458, 3035, 2972, 1738, 1363, 1222; 1H-NMR (400 MHz, DMSO-d6): δ 8.33 (bs, NH), 7.51–7.22 (m, 11H, CH & CH & 2Ph), 2.94 (q, 4H, J = 7.3 Hz, CH2CH3), 1.16 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 203.2, 182.7, 136.3, 135.5, 135.4, 130.1, 130.0, 129.5, 128.1, 127.7, 124.7, 41.9, 11.6; LC/MS (ESI): 404 [M]+; anal. for C20H24N2OS3; calcd: C, 59.37; H, 5.98; N, 6.92; found: C, 59.40; H, 6.01; N, 6.90.%
:1) (4a·HNEt2). 4a·HNEt2 was prepared from 2-thioxothiazolidin-4-one 1, benzaldehyde and thiophenol according to the general procedure (GP1) yielding yellow crystalline materials (1.17 g, 2.9 mmol, 96%). m.p: 180 °C; IR (KBr, cm−1): 3458, 3035, 2972, 1738, 1363, 1222; 1H-NMR (400 MHz, DMSO-d6): δ 8.33 (bs, NH), 7.51–7.22 (m, 11H, CH & CH & 2Ph), 2.94 (q, 4H, J = 7.3 Hz, CH2CH3), 1.16 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 203.2, 182.7, 136.3, 135.5, 135.4, 130.1, 130.0, 129.5, 128.1, 127.7, 124.7, 41.9, 11.6; LC/MS (ESI): 404 [M]+; anal. for C20H24N2OS3; calcd: C, 59.37; H, 5.98; N, 6.92; found: C, 59.40; H, 6.01; N, 6.90.%The structure of 4a·HNEt2 was confirmed by X-ray crystal structure analysis. A crystal suitable for X-ray analysis of the compound formed in DCM/EtOH at room temperature gives yellow crystal.†
:1) (4c·HNEt2). 4c·HNEt2 was prepared from 2-thioxothiazolidin-4-one 1, p-chlorobenzaldehyde and thiophenol according to the general procedure (GP1) yielding yellow powder (1.21 g, 2.76 mmol, 92%). m.p: 103 °C; IR (KBr, cm−1): 3459, 3035, 2973, 1738, 1365, 1226; 1H-NMR (400 MHz, DMSO-d6): δ 8.34 (bs, 1H, NH), 7.52 (d, 2H, J = 8.0 Hz, Ph), 7.38 (d, 2H, J = 8.0 Hz, CH & CH), 7.36 (d, 2H, J = 8.0 Hz, Ph), 7.30–7.21 (m, 5H, Ph), 2.94 (q, 2H, J = 6.6 Hz, CH2CH3), 1.16 (t, 3H, J = 6.6 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 202.6, 182.7, 136.3, 136.1, 134.4, 133.6, 131.7, 130.0, 129.8, 129.6, 128.1, 127.7, 123.4, 41.9, 11.6; LC/MS (ESI): 439 [M]+; anal. for C20H23ClN2OS3; calcd: C, 54.71; H, 5.28; N, 6.38; found: C, 54.70; H, 5.25; N, 6.37.%:1) (4d·HNEt2). 4d·HNEt2 was prepared from 2-thioxothiazolidin-4-one 1, p-bromobenzaldehyde and thiophenol according to the general procedure (GP1) yielding yellow needle (931 mg, 2.7 mmol, 90%). m.p: 240 °C; IR (KBr, cm−1): 3458, 3015, 2970, 1738, 1365, 1227; 1H-NMR (400 MHz, DMSO-d6): δ 8.30 (bs, 1H, NH), 7.69 (d, 2H, J = 8.0 Hz, Ph), 7.53 (d, 2H, J = 8.0 Hz, CH & CH), 7.49 (d, 2H, J = 8.0 Hz, Ph), 7.40–7.22 (m, 5H, Ph), 2.92 (q, 2H, J = 6.6 Hz, CH2CH3), 1.16 (t, 3H, J = 6.6 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 200.2, 167.1, 134.1, 134.1, 132.6, 132.2, 130.0, 128.1, 127.7, 125.9, 123.2, 41.5, 11.6; LC/MS (ESI): 345 [M]+; anal. for C20H23BrN2OS3; calcd: C, 49.68; H, 4.79; N, 5.79; found: C, 49.70; H, 4.76; N, 5.81.%:1) (4h·HNEt2). 4h·HNEt2 was prepared from 2-thioxothiazolidin-4-one 1,2-naphthaldehyde and thiophenol according to the general procedure (GP1) as yellow powder (1.24 g, 2.73 mmol, 91%). m.p: 160 °C; IR (KBr, cm−1): 3458, 3031, 2970, 1738, 1663, 1348, 1216; 1H-NMR (400 MHz, DMSO-d6): δ 10.40 (s, NH), 9.16 (d, 1H, J = 8.0 Hz, naphthyl) 8.27–7.29 (m, 13H, CH, CH, naphthyl, Ph), 2.92 (q, 2H, J = 6.6 Hz, CH2CH3), 1.16 (t, 3H, J = 6.6 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 198.7, 194.9, 137.3, 136.3, 135.8, 133.8, 132.1, 131.6, 131.4, 131.2, 131.1, 130.3, 130.0, 129.5, 129.4, 129.2, 128.1, 127.9, 127.7, 127.4, 127.2, 126.2, 125.9, 124.6, 123.8, 41.7, 11.6; LC/MS (ESI): 454 [M]+; anal. for C24H26N2OS3; calcd: C, 63.40; H, 5.76; N, 6.16; found: C, 63.43; H, 5.75; N, 6.15.%:1) (4i·HNEt2). 4i·HNEt2 was prepared from 2-thioxothiazolidin-4-one 1,2,4-dichlorobenazaldehyde and thiophenol according to the general procedure (GP1) yielding yellow powder (1.16 g, 2.46 mmol, 82%). m.p: 85 °C; IR (KBr, cm−1): 3458, 3022, 2970, 1738, 1348, 1216; 1H-NMR (400 MHz, DMSO-d6): δ 8.37 (bs, 1H, NH), 7.72 (s, 1H, Ph), 7.61–7.27 (m, 9H, CH & CH & Ph), 2.92 (q, 2H, J = 6.6 Hz, CH2CH3), 1.16 (t, 3H, J = 6.6 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 202.1, 181.7, 138.8, 136.3, 135.4, 134.1, 132.3, 130.6, 130.0, 129.5, 128.9, 128.6, 128.1, 128.0, 127.7, 118.6, 41.9, 11.5; LC/MS (ESI): 473 [M]+; anal. for C20H22Cl2N2OS3; calcd: C, 50.73; H, 4.68; Cl, 14.97; N, 5.92; found: C, 50.72; H, 4.70; Cl, 5.01; N, 5.90.%:1) (4k·HNEt2). 4k·HNEt2 was prepared from 2-thioxothiazolidin-4-one 1,3-methylbenzaldehyde and thiophenol according to the general procedure (GP1) yielding yellow powder (1.11 g, 2.67 mmol, 89%). m.p: 145 °C; IR (KBr, cm−1): 3458, 3035, 2972, 1738, 1363, 1222; 1H-NMR (400 MHz, CDCl3): δ 8.30 (bs, NH), 7.49 (d, 2H, J = 7.3 Hz, Ph),7.39 (s, 1H, Ph),7.31–7.11 (m, 8H, CH & CH & Ph), 3.14 (q, 4H, J = 7.3 Hz, CH2CH3),2.31 (s, 3H, CH3), 1.36 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, CDCl3): δ = 207.0, 182.6, 138.8, 134.5, 132.7, 130.9, 130.5, 129.4, 129.1, 129.0, 128.7, 127.6, 127.2, 125.6, 42.8, 21.5, 11.6; LC/MS (ESI): 418 [M]+; anal. for C21H26N2OS3; calcd: C, 60.25; H, 6.26; N, 6.69; found: C, 60.22; H, 6.25; N, 6.70.%General procedure for the synthesis of 3·HNEt2 (GP3)
A mixture of aldehyde 2 (3 mmol, 318 mg), 2-thioxothiazolidin-4-one 1 (3 mmol, 400 mg), and Et2NH (3 mmol, 310 μl) in 3 ml of degassed H2O was stirred at room temperature for 3–5 hours until TLC showed complete disappearance of the reactants. The product precipitated and the mixture was filtered and the solid portion was recrystallized to obtain the pure product 3·HNEt2.:1) (3a·HNEt2). 3a·HNEt2 was prepared from 2-thioxothiazolidin-4-one 1, and benzaldehyde according to general procedure (GP3) as yellow crystalline materials; IR (KBr, cm−1): 3458, 3016, 2970, 1738, 1430, 1366, 1228; 1H-NMR (400 MHz, DMSO-d6): δ 8.38 (bs, 1H, NH), 7.55–7.47 (m, 5H, Ph), 7.41 (s, 1H, CH), 2.95 (q, 4H, J = 13.9 Hz, CH2CH3), 1.16 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 200.1, 177.3, 134.6, 130.5, 130.1, 129.7, 128.0, 41.9, 11.6; LC/MS (ESI): 294 [M]+; anal. for C14H18N2OS2; calcd: C, 57.11; H, 6.16; N, 9.51; found: C, 57.12; H, 6.16; N, 9.50.%The structure 3a·HNEt2 was confirmed by X-ray crystal structure analysis. A crystal suitable for X-ray analysis of the compound formed in DCM/EtOH at room temperature gives yellow crystal.†
:1) (3b·HNiPr2). 3b·HN·iPr2 was prepared from 2-thioxothiazolidin-4-one 1, and benzaldehyde according to the general procedure (GP3) using diisopropylamine to yield yellow crystalline materials; IR (KBr, cm−1): 3454, 3028, 2970, 1738, 1430, 1366, 1225; 1H-NMR (400 MHz, DMSO-d6): δ 8.38 (bs, 1H, NH), 7.51–7.44 (m, 5H, Ph), 7.18 (s, 1H, CH), 3.35 (m, 1H, CH(CH3)2), 1.20 (d, 6H, J = 6.6 Hz, CH(CH3)2); 13C-NMR (100 MHz, DMSO-d6): δ = 203.8, 184.4, 136.2, 135.7, 130.1, 129.5, 128.9, 124.0, 47.7, 19.3; LC/MS (ESI): 322 [M]+; anal. for C16H22N2OS2; calcd: C, 59.59; H, 6.88; N, 8.69; found: C, 59.60; H, 6.86; N, 8.71.%The structure 3b·HN·iPr2 was confirmed by X-ray crystal structure analysis. A crystal suitable for X-ray analysis of the compound formed in DCM/EtOH at room temperature was used in the structure determination.†
:1) (3c·HNEt2). 3c·HNEt2 was prepared from 2-thioxothiazolidin-4-one 1, and p-chlorobenzaldehyde according to general procedure (GP3) to yield yellow crystalline materials; IR (KBr, cm−1): 3458, 3016, 2970, 1738, 1430, 1366, 1228; 1H-NMR (400 MHz, DMSO-d6): δ 8.40 (bs, 1H, NH), 7.53–7.24 (m, 5H, Ph & CH), 2.94 (q, 4H, J = 13.9 Hz, CH2CH3), 1.16 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 196.6, 171.2, 132.6, 132.5, 130.0, 129.9, 41.9, 11.6; LC/MS (ESI): 328 [M]+; anal. for C14H17ClN2OS2; calcd: C, 51.13; H, 5.21; N, 8.52; found: C, 51.12; H, 5.20; N, 8.50.%The structure 3c·HNEt2 was confirmed by X-ray crystal structure analysis. A crystal suitable for X-ray analysis of the compound formed in DCM/Et2O at room temperature was used for structure determination.†
General procedure for the synthesis of 3d–g (GP4)
A mixture of aldehyde 2 (3 mmol, 318 mg), 2-thioxothiazolidin-4-one 1 (3 mmol, 400 mg), and Et2NH (3 mmol, 310 μl) in 3 ml of degassed H2O was stirred at room temperature for 3–5 hours until TLC showed complete disappearance of the reactants. Water (3 ml) was the added into the reaction mixture, then extracted with DCM/EtOH (3 × 50 ml) and the organic phase washed with 10% HCl (2 × 50 ml), followed by brine (2 × 50 ml), and dried over MgSO4, and filtered off and evaporated to afford 3d–g.%), 7.68 (d, 1H, J = 2.9 Hz, thiophene), 7.28 (t, 1H, J = 3.6 Hz, thiophene); 13C-NMR (100 MHz, DMSO-d6): δ = 195.1, 169.5, 137.9, 135.9, 134.8, 129.8, 125.3, 123.5; LC/MS (ESI): 227 [M]+; anal. for C8H5NOS3; calcd: C, 42.27; H, 2.22; N, 6.16; found: C, 42.28; H, 2.20; N, 6.17.%:1) (3e). 3e was prepared from 2-thioxothiazolidin-4-one 1, and furan-2-carbaldehyde according to the general procedure (GP4) as brown powder (620 mg, 2.73 mmol, 91%). m.p: 150 °C; IR (KBr, cm−1): 3458, 3016, 2970, 1738, 1430, 1366, 1228; 1H-NMR (400 MHz, DMSO-d6): δ 13.67 (bs, 1H, SH), 8.1 (s, 1H, NH), 7.52 (d, 1H, J = 7.3 Hz, furan), 7.47 (s, 1H, CH), 7.38 (t, 1H, J = 7.3 Hz, furan, major isomer), 7.29 (t, 1H, J = 7.3 Hz, furan, minor isomer), 7.16 (d, 1H, J = 2.9 Hz, furan), 6.76 (bs, 1H, furan),; 13C-NMR (100 MHz, DMSO-d63): δ = 197.7, 169.7, 150.0, 148.8, 136.3, 130.0, 128.1, 127.7, 123.2, 120.3, 118.1, 114.4; LC/MS (ESI): 210 [M]+; anal. for C8H5NO2S2; calcd: C, 45.48; H, 2.39; N, 6.63; found: C, 45.44; H, 2.42; N, 6.61.%), 7.28 (bs, 1H, Pyrrol), 6.52 (bs, 1H, Pyrrol), 6.39 (bs, 1H, Pyrrol); 13C-NMR (100 MHz, DMSO-d6): δ = 195.3, 169.7, 127.7, 126.3, 122.5, 117.4, 115.5, 113.3; LC/MS (ESI): 210 [M]+; anal. for C8H6N2OS2; calcd: C, 45.69; H, 2.88; N, 13.32; found: C, 45.72; H, 2.90; N, 13.33.%), 7.43 (t, 1H, J = 1.9 Hz, Pyridine); 13C-NMR (100 MHz, DMSO-d6): δ = 202.5, 169.8, 151.6, 150.0, 138.1, 130.2, 128.7, 127.9, 124.5; LC/MS (ESI): 222 [M]+; anal. for C9H6N2OS2; calcd: C, 48.63; H, 2.72; N, 12.60; found: C, 48.62; H, 2.72; N, 12.61.%Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the research group project Number RGP-VPP-257.Notes and references
- For reviews, see: (a) R. B. Lesyk and B. S. Zimenkovesky, Curr. Org. Chem., 2004, 8, 1547 CrossRef CAS; (b) L. P. Masic and T. Tomasic, Curr. Med. Chem., 2009, 16, 1596 CrossRef.
- T. T. Talele, P. Arora, S. S. Kulkarni, M. R. Patel, S. Singh, M. Chudayeu and N. Kaushik-Basu, Bioorg. Med. Chem., 2010, 18, 4630 CrossRef CAS PubMed.
- C. A. Whitesitt, R. L. Simon, J. K. Reel, S. K. Sigmund, M. L. Phillips, J. K. Shadle, L. J. Heinz, G. A. Koppel, D. C. Hunden, S. L. Lifer, D. Berry, J. Ray, S. P. Little, X. Liu, W. S. Marshall and J. A. Panetta, Bioorg. Med. Chem. Lett., 1996, 6, 2157 CrossRef CAS.
- (a) D. Havrylyuk, L. Mosula, B. Zimenkovsky, O. Vasylenko, A. Gzella and R. Lesyk, Eur. J. Med. Chem., 2010, 45, 5012 CrossRef CAS PubMed; (b) T. Tihomir, Z. Nace, M. P. Manica, K. Danijel and P. M. Lucija, Eur. J. Med. Chem., 2010, 45, 1667 CrossRef PubMed; (c) R. Murugan, S. Anbazhagan and S. S. Narayanan, Eur. J. Med. Chem., 2009, 44, 3272 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Joshi, C. Vargas, P. Boisguerin, A. Diehl, G. Krause, P. Schmieder, K. Moelling, V. Hagen, M. Schade and H. Oschkinat, Angew. Chem., Int. Ed., 2006, 45, 3790 CrossRef CAS PubMed; (b) J. P. Powers, D. E. Piper, Y. Li, V. Mayorga, J. Anzola, J. M. Chen, J. C. Jaen, G. Lee, J. Liu, M. G. Peterson, G. R. Tonn, Q. Ye, N. P. C. Walker and Z. Wang, J. Med. Chem., 2006, 49, 1034 CrossRef CAS PubMed; (c) A. M. Gilbert, M. G. Bursavich, S. Lombardi, K. E. Georgiadis, E. Reifenberg, C. R. Flannery and E. A. Morris, Bioorg. Med. Chem. Lett., 2007, 17, 1189 CrossRef CAS PubMed; (d) N. D. Sonawane and A. S. Verkman, Bioorg. Med. Chem., 2008, 16, 8187 CrossRef CAS PubMed; (e) N. Zidar, T. Tomašič, R. Šink, V. Rupnik, A. Kovač, S. Turk, D. Patin, D. Blanot, C. C. Martel, A. Dessen, M. M. Premru, A. Zega, S. Gobec, L. P. Mašič and D. Kikelj, J. Med. Chem., 2010, 53, 6584 CrossRef CAS PubMed; (f) B. R. Prashantha Kumar and M. J. Nanjan, Bioorg. Med. Chem. Lett., 2010, 20, 1953 CrossRef PubMed; (g) T. Sohda, K. Mizuno and Y. Kawamatsu, Chem. Pharm. Bull., 1984, 32, 4460 CrossRef CAS.
- M. Pulici and F. Quartieri, Tetrahedron Lett., 2005, 46, 2387 CrossRef CAS PubMed.
- P. T. Anastas and J. Warner, Green Chemistry. Theory and Practice, Oxford University Press, New York, 1998, p. 30 Search PubMed.
- (a) C. Bhanja, S. Jena, S. Nayak and S. Mohapatra, Beilstein J. Org. Chem., 2012, 8, 1668 CrossRef CAS PubMed; (b) J. Clayden and P. MacLellan, Beilstein J. Org. Chem., 2011, 7, 582 CrossRef CAS PubMed; (c) S. Kanemasa and K. Ito, Eur. J. Org. Chem., 2004, 4741 CAS; (d) K. Nishimura and K. Tomioka, Yakugaku Zasshi, 2003, 123, 9 CrossRef CAS.
- (a) M. Saito, M. Nakajima and S. Hashimoto, Tetrahedron, 2000, 56, 9589 CrossRef CAS; (b) M. Bandini, P. G. Cozzi, M. Giacomini, P. Melchiorre, S. Selva and A. Umani-Ronchi, J. Org. Chem., 2002, 67, 3700 CrossRef CAS PubMed; (c) S. K. Garg, R. Kumar and A. K. Chakraborti, Synlett, 2005, 1370 CAS; (d) A. T. Khan, S. Ghosh and L. H. Choudhury, Eur. J. Org. Chem., 2006, 2226 CrossRef CAS; (e) S. Banerjee, J. Das, R. P. Alvareza and S. Santra, New J. Chem., 2010, 34, 302 RSC; (f) J. S. Yadav, B. V. S. Reddy and G. Baishya, J. Org. Chem., 2003, 68, 7098 CrossRef CAS PubMed; (g) B. C. Ranu, S. S. Dey and A. Hajra, Tetrahedron, 2003, 59, 2417 CrossRef CAS; (h) B. C. Ranu and S. S. Dey, Tetrahedron, 2004, 60, 4183 CrossRef CAS PubMed.
- M. S. Abaee, S. Cheraghi, S. Navidipoor, M. M. Mojtahedi and S. Forghani, Tetrahedron Lett., 2012, 53, 4405 CrossRef CAS PubMed.
- (a) A. Barakat, A. M. A. Al-Majid, M. Shahidul Islam and Z. A. Al-Othman, Tetrahedron, 2013, 69, 5185 CrossRef CAS PubMed; (b) A. Barakat and A. M. A. Al-Majid, Arabian J. Chem., 2013 DOI:10.1016/j.arabjc.2012.12.025; (c) A. M. A. Al-Majid, A. Barakat, Y. N. Mabkhot and M. Shahidul Islam, Int. J. Mol. Sci., 2012, 13, 2727 CrossRef CAS PubMed; (d) A. M. A. Al-Majid, A. Barakat, H. J. Al-Najjar, Y. N. Mabkhot, H. A. Ghabbour and H-K. Fun, Int. J. Mol. Sci., 2013, 14, 23762 CrossRef PubMed.
- For selected examples, see: (a) M. Zhang, C. D. Wang, S. L. Yu, Z. B. Tian and L. Zhang, Chemical Journal of Chinese Universities, 1994, 15, 1647 (Chem. Abstr., 1995, 122, 213992z) CAS; (b) A. B. Alloum, S. Bakkas, K. Bougrin and M. Soufiaoui, New J. Chem., 1998, 22, 809 RSC; (c) F. Toda, K. Tanaka and K. Hamai, J. Chem. Soc., Perkin Trans. 1, 1990, 320 Search PubMed; (d) C. J. Li, Chem. Rev., 1993, 93, 2023 CrossRef CAS; (e) S. Deshayes, M. Liagre, A. Loupy, J. L. Luche and A. Petit, Tetrahedron, 1999, 55, 10851 CrossRef CAS; (f) C. L. Lee and M. M. Sim, Tetrahedron Lett., 2000, 41, 5729 CrossRef CAS.
- (a) M. Gruttadauria, F. Giacalone, A. M. Marculesco, P. L. Meo, S. Riela and R. Noto, Eur. J. Org. Chem., 2007, 4688 CrossRef CAS; (b) R. Breslow, Acc. Chem. Res., 2004, 37, 471 CrossRef CAS PubMed; (c) D. G. Blackmond, A. Armstrong, V. Coombe and A. Wells, Angew. Chem., Int. Ed. Engl., 2007, 46, 3798 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 930797, 931815, 930934 and 930932. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra46551a |
| This journal is © The Royal Society of Chemistry 2014 |