New clicked thiirane derivatives as gelatinase inhibitors: the relevance of the P1′ segment†
B. Fabrea,
K. Filipiakab,
C. Codercha,
J. M. Zapicoa,
Rodrigo J. Carbajoc,
Anne K. Schottc,
Antonio Pineda-Lucenac,
B. de Pascual-Teresa*a and
A. Ramos*a
aDepartamento de Química y Bioquímica, Facultad de Farmacia, Universidad CEU San Pablo, Urbanización Monteprincipe, 28668, Boadilla del Monte, Madrid, Spain. E-mail: aramgon@ceu.es
bDepartment of Molecular Biology, Faculty of Biotechnology and Environment Sciences, The John Paul II Catholic University of Lublin, 20-718, Lublin, Poland. E-mail: bpaster@ceu.es
cStructural Biochemistry Laboratory, Advanced Therapies Program, Centro de Investigación Príncipe Felipe, C/Eduardo Primo Yúfera 3, 46012 Valencia, Spain
First published on 3rd April 2014
Abstract
Gelatinases (MMP-2 and MMP-9), a subfamily of Matrix Metalloproteinases (MMPs), are involved in several pathologies and especially in cancer. Thiirane is a latent-zinc binding group used for the design of potent inhibitors of gelatinases. Here we report a new family of thiirane inhibitors, obtained by click chemistry. Thus, an azide fragment containing the thiirane group was connected to several lipophilic alkynes, which were designed to interact with the S1′ pocket of the two gelatinases. Our hit compound (2f) displayed submicromolar inhibition of MMP-2 (IC50 = 0.62 μM). Computational studies have been used to compare the binding mode of compound 2f in MMP-2 with the reference thiirane inhibitor (SB-3CT), allowing us to discuss the relevance of the P1′ segment in order to maximize potency.
Introduction
Gelatinases (MMP-2 and MMP-9) belong to the matrix metalloprotease family (MMP), a group of zinc-dependent endopeptidases.1 In the 90 s, MMPs were considered as ideal targets, mainly for cancer and inflammation diseases, due to their ability to degrade extracellular matrix (ECM) components such as collagen.2 However, their role is more complicated than first thought, and MMPs have been demonstrated to participate also in the activation, regulation or destruction of signalling molecules, such as growth factors and cytokines.3,4 Furthermore, related to their implication in cancer, some MMPs play protective roles depending on the cancer localization or stage.5 This, in addition to the dose-limiting musculoskeletal syndrome (MSS), is one of the reasons for the disappointing results obtained in clinical trials with broad-spectrum MMP inhibitors (MMPIs).6,7 Thus, medicinal chemists from the MMP field are now looking for more selective inhibitors within the MMP family. This is mainly achieved by exploring differences in the lipophilic S1′ pocket of MMPs, also known as the selectivity pocket,8 using compounds including or not a group capable to bind the catalytic zinc ion (zinc binding group or ZBG).9–11The hydroxamate moiety was largely used as ZBG for potent inhibition of MMPs.11,12 However, hydroxamates suffer from metabolic liability, toxicity and generally low selectivity. For this reason, non-hydroxamate ZBGs have been developed, such as carboxylates, thiolates, phosphonates, phosphinic derivatives, hydroxypyrone derivatives and pyrimidinetriones, among others.10 More than a decade ago, Brown et al.13 reported thiirane SB-3CT as a new slow-binding inhibitor, that displayed remarkable selective inhibition of gelatinases. Additionally, SB-3CT efficiently inhibits cancer growth and metastasis in liver and bone murine models.14,15 The proposed inhibition mechanism (Fig. 1), based on experimental and computational studies, implies the opening of SB-3CT thiirane ring within the MMP catalytic cleft, leading to the real active specie 1.16–19 As SB-3CT displays high metabolism – in particular at the phenoxyphenyl part – and poor aqueous solubility,20–24 Mobashery et al. replaced the distal phenoxy group of SB-3CT by triazole fragments. However only weak gelatinases inhibitors were obtained.25
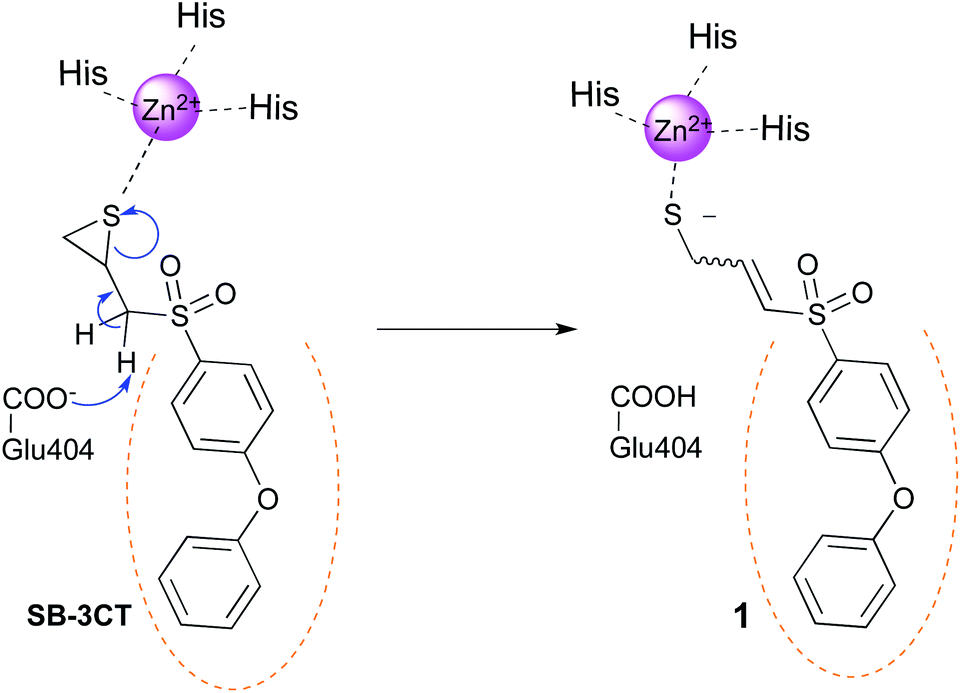 | ||
| Fig. 1 Proposed mechanism for the slow inhibition of MMPs by SB-3CT. The catalytic Glu404 removes a proton from the inhibitor, leading to the opening of the thiirane ring. The catalytic zinc ion of the MMP is represented in magenta and the S1′ pocket is symbolized by orange dots. Numbering of the full proMMP-2 is used in this work.26 | ||
We previously reported a series of highly potent triazole based MMP-2 inhibitors with hydroxamate as ZBG obtained following the click-chemistry approach,27,28 in which an azide containing segment was connected to a series of alkynes (named P1′ fragments according to Schechter and Berger nomenclature29) designed to interact with the lipophilic residues of the S1′ pocket. This work led to inhibitors with high selectivity for MMP-2 over MMP-9.30 Based on those encouraging results, we decided to replace the hydroxamate ZBG for the reasons stated above. Being interested in the inhibition of MMP-2 because of its potential role in cancer,31 we chose the thiirane latent ZBG as thiirane derivatives displayed good selectivity for this enzyme.13,32 Here we report the discovery of a hit compound (2f) that showed submicromolar activity against MMP-2, thus improving the potency of the clicked thiirane derivatives previously reported by Mobashery group in their attempt to improve the metabolic behaviour of SB-3CT.25 To design our compounds, we connected the azide core containing the latent-ZBG to the series of alkynes that provided the most potent inhibitors in our previous work,27 as well as to two new alkyne fragments (Fig. 2). Docking studies and molecular dynamics (MD) experiments have been performed in order to rationalize the difference of inhibitory activity between 2f and the reference compound SB-3CT. This work allowed us to discuss the importance of the P1′ fragment in the potency of this type of inhibitors.
 | ||
| Fig. 2 Click-chemistry approach for the synthesis of 2. | ||
Results and discussion
Chemistry
Compounds 2a–f were synthesized as depicted in Scheme 1. As the chirality of SB-3CT has no influence on its biological action on gelatinases,33 – the two enantiomers of SB-3CT and the racemic mixture of both displayed similar kon and koff in vitro – we synthesised the racemic mixtures of 2a–f. To obtain the α-sulfone thiirane moiety, we used the methodology developed by Mobashery et al.23 An azide group was introduced to allow the rapid growth of the molecule by coupling with fragments designed to interact with gelatinases S1′ pocket by means of click chemistry. The alkynes used here were chosen based on our previous work in this field.27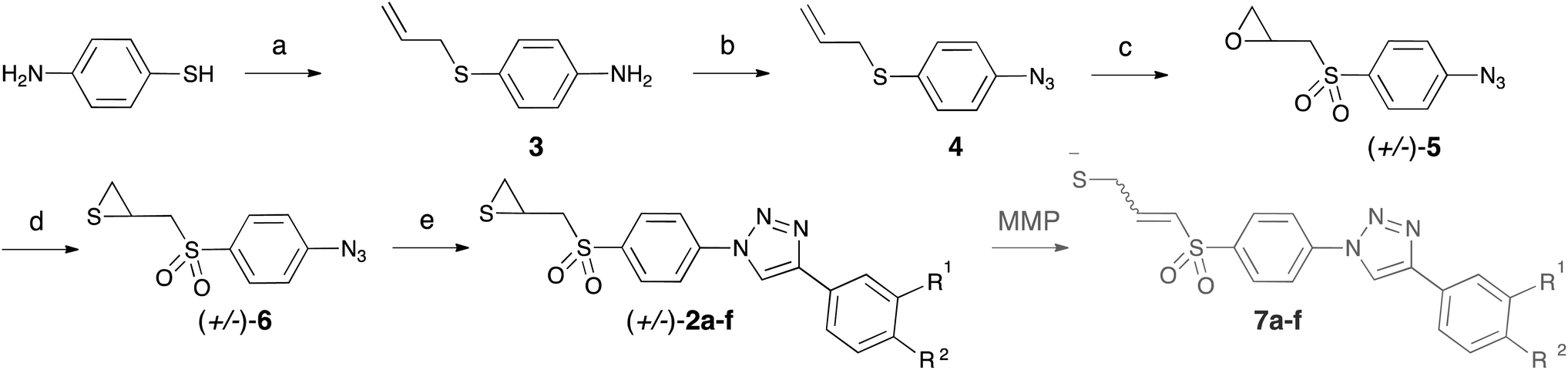 | ||
Scheme 1 Synthesis of 2a–f. Reagents and conditions: (a) NaH at 0 °C followed by allyl chloride, DMF, 68%; (b) tBuONO, TMSN3, CH3CN, 84%; (c) mCPBA (excess), CH2Cl2, 37%; (d) thiourea, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH–CH2Cl2, 81%; (e) CuSO4·5H2O (0.06 eq.), sodium ascorbate (0.20 eq.), alkyne. :1 MeOH–CH2Cl2, 81%; (e) CuSO4·5H2O (0.06 eq.), sodium ascorbate (0.20 eq.), alkyne. | ||
Selective S-alkylation of 4-aminothiophenol by allyl chloride provided compound 3. Less than 20% of the starting material was converted into the corresponding N-alkylated by-product, which was easily separated from the S-alkylated product by column chromatography. Compound 3 was treated with tert-butyl nitrite to afford the diazonium salt, which was reacted in situ with azidotrimethylsilane as azide source, to form azide 4. Oxidation with mCPBA led to α-sulfone epoxide 5. In this step, an excess of mCPBA was necessary, and the reaction was stopped after one week, before total completion. Other oxidation agents (peracetic acid and hydrogen peroxide) were tried, as well as different temperatures of reaction, without any improvement in the final yield. Epoxide 5 was converted into thiirane 6 by reaction with thiourea. From the azide 6, the triazole family 2a–f was obtained by mean of copper catalysed azide–alkyne cycloadditions (CuAAC), using copper sulphate pentahydrate and sodium ascorbate as catalytic system. Three solvents (1:1H2O–tBuOH, MeCN and DMF) were screened for this transformation, DMF giving the best results.
The alkynes necessary for the synthesis of 2a–e are commercially available, whereas the alkyne necessary for the synthesis of 2f was synthesized according to the literature procedure.34 In Scheme 1, we also show the results of the opening of thiiranes 2a–f to form allyl thiolates 7a–f, expected to occur in the protein catalytic centre.
Biology
Compounds activities against MMP-2 and MMP-9 were evaluated in vitro, using a colorimetric assay.The reaction was measured both after 45 min and 2 h of incubation of the enzyme with the inhibitors (Table 1). Thiiranes are slow-binding inhibitors of gelatinases, and they need a previous enzymatic transformation to the corresponding allyl thiolate, which is the real inhibitor (see mechanism in Fig. 1). The higher activity observed by us after a longer incubation period (2 h) is in accordance with the described mechanism of action of thiirane inhibitors. The most active compound is 2f (IC50 = 0.62 μM against MMP-2 and IC50 = 2.71 μM against MMP-9), with a 4-pyridyl substituent in the terminal aromatic ring. The replacement of the 4-phenoxy segment in SB-3CT by a triazole, using a CuAAC approach, is thus a useful strategy to attain structural diversity in this type of inhibitors.
|
|||||
|---|---|---|---|---|---|
| Cpd | R1 | R2 | MMP-2 | MMP-9 | |
| 45b min | 2b h | 2b h | |||
| a Determined by a colorimetric assay. Enzymatic data are mean values from three independent experiments. SD are within ±10%.b Incubation times.c Remaining activity (%) at 10 μM.d Remaining activity (%) at 20 μM. | |||||
| 6 | — | — | >50 | >50 | >50 |
| 2a | H | N(CH3)2 | >20 | >20 | >20 |
| 2b | H | (CH2)4CH3 | >20 | n.d. | n.d. |
| 2c | H | C6H5 | 44.4%c | 2.36 | 12.04 |
| 2d | H | OCH3 | >20 | 87.5%d | >20 |
| 2e | F | H | ∼20 | 10.9 | 79.9%d |
| 2f | H |  |
46.8%c | 0.62 | 2.71 |

Substitution of the pyridyl ring present in 2f by phenyl (2c) and m-F (2e) brought about a decrease in activity against MMP-2 and MMP-9. Finally, the introduction of N,N-dimethylamino, n-pentyl or methoxy groups gave inactive compounds against both metalloproteinases at 20 μM.
In a similar attempt to apply click chemistry to the synthesis of thiirane-based gelatinase inhibitors by Mobashery's group,25 they obtained compounds 8 (Fig. 3) that showed to be weak inhibitors of MMP-2 (in general, more than 50% of remaining MMP-2 activity at 30 μM final concentration and after 3 h of incubation). The authors suggested that the presence of a two atoms link (–CH2O–) between the triazole and phenyl rings could be the cause of the lack of activity. The good inhibitory activity observed for 2f, where the triazole is directly connected to the phenyl is in complete accordance with this hypothesis.
 | ||
| Fig. 3 Clicked-thiiranes synthesized by Mobashery et al.25 | ||
Here it should be stressed that direct SAR analysis is rather perilous. The binding of these thiirane derivatives could be divided in three steps: (1) binding to the MMP; (2) hydrogen abstraction by Glu404 and subsequent opening of the thiirane ring (see Fig. 1); (3) binding of the resulting open compound and chelation of the catalytic zinc ion by the thiolate moiety. Classical SAR studies usually limit to step one, but here the structure of the inhibitor may importantly influence steps two and three. We thus carried out computational studies to examine the influence of the P1′ fragment on the inhibitor potency.
Binding to MMP-2: NMR and computational studies
Given the difference in potency of our inhibitors with the reference thiirane SB-3CT, we verified if our compounds bind the MMP-2 catalytic cleft by NMR intermolecular-interaction experiments. We have previously used the waterLOGSY experiment35 in the characterization of novel inhibitors binding MMP-2.27 Here we have applied a similar methodology to the study of the interaction between MMP-2 and compounds 6, 2a–f. In general, compounds 2a–f show very low solubility in water. As a result, compounds 2a–d and 2f did not show recognizable resonances in the regular 1D 1H NMR spectra recorded in the protein buffer (see Materials & methods for further details) even with the addition of supplementary DMSO (3%). Likewise, no significant information could be extracted from their waterLOGSY experiments with MMP-2. However, compounds 6 and 2e were partially soluble under the NMR experimental conditions, both showing positive intermolecular binding with MMP-2 in the waterLOGSY spectra. To evaluate if such binding takes place at the S1′ pocket, waterLOGSY competition experiments were repeated in the presence of 2R-[(4-biphenylsulfonyl)amino]-N-hydroxy-3-phenylpropinamide (BiPS), a commercially available MMP2/9 inhibitor (IC50 (MMP-2): 17 nM; IC50 (MMP-9): 30 nM). The addition of BiPS to the MMP-2/6 mixture yielded positive interaction for both compounds in the waterLOGSY, with no significant reduction of signals from compound 6 compared to the MMP-2/6 waterLOGSY spectrum. The inverse competition experiment (1st: waterLOGSY MMP-2/BiPS; 2nd: addition of 6, waterLOGSY MMP-2/BiPS/6) gave analogous results, suggesting that BiPS and compound 6 do not compete for the same binding site of the protein. In the case of compound 2e, the addition of BiPS to the MMP-2/2e sample yielded positive binding for BiPS and no interaction for 2e, indicating that BiPS displaces 2e from the binding pocket due to its higher affinity for the protein (ESI figure†). This result was confirmed by the inverse waterLOGSY competition experiment, where the positive signals of the interaction BiPS – MMP-2 were not affected by the addition of compound 2e. These results indicate that compound 2e binds MMP-2 at the S1′ pocket.Docking studies
Docking simulations in MMP-2 catalytic centre were carried out for 2a–f and SB-3CT for comparison.As seen before, thiirane derivatives are slow binding inhibitors due to the opening of the thiirane ring within the MMP-2 catalytic cleft. For the docking studies, we considered both open and closed forms to study the ability of the thiiranes, as well as the corresponding allyl thiolates, to correctly bind to the MMP-2 catalytic centre. For each form (closed and open), each stereoisomer (S or R, Z or E) was considered. Indeed, using QM/MM calculations, Mobashery et al. proposed that (R)-SB-3CT leads preferentially to the allyl thiolate with E configuration,18 whereas (S)-SB-3CT leads to the Z isomer.36
According to the literature,18 three important interactions could be expected for those compounds: (1) hydrophobic interactions at the S1′ pocket; (2) H-bond(s) between one sulfone oxygen of the inhibitor and Leu191 and/or Ala192 backbone; (3) interaction between the sulphur atom and the catalytic zinc ion, although such coordination was only demonstrated experimentally for the allyl thiolate.
We carried out the docking studies as explained in the Experimental section.
• Docking of thiirane derivatives 2a–f.
In a first attempt, dockings were performed with 1CK7 structure, the same used to study the mechanism of SB-3CT.18 While SB-3CT gave correct docking poses (that is to say with the three interaction types listed above), 2a–f didn't. One possible reason for this is that in 1CK7 structure no inhibitor is present, leading to a tight S1′ pocket, that cannot accommodate the long P1′ fragments used here (we previously observed false positive docking results with 1CK7 structure for inhibitors with long/bulky P1′ segment). Consequently, dockings were repeated using a structure extracted from a MD trajectory of the catalytic domain of 1CK7 bound to a hydroxamate inhibitor with a similar P1′ fragment than those used here.28 Surprisingly, no correct binding for SB-3CT was observed in this model. The curved biphenyl ether moiety could not fit into the S1′ pocket together with a good coordination of the zinc ion by the sulphur atom. By contrast, in the most energetically favoured binding modes, 2a–f displayed simultaneously the three interactions listed above. Fig. 4 shows the binding modes of (R)- and (S)-2f in this MMP-2 structure.
 | ||
| Fig. 4 Most energetically favoured binding mode for (R)-2f (A) and (S)-2f (B) in MMP-2. The hydrogens from the ligand that are transferred to Glu404 during the thiirane opening are depicted. H-bonds are represented by orange dots. Zn2+–sulphur distances (blue dots) are 2.62 Å (A) and 2.72 Å (B), respectively. | ||
The binding modes of both enantiomers are similar in all compounds, what is in consonance with the results described for (S)- and (R)-SB-3CT.33 It is also important to note that the hydrogen that is taken by Glu404 in the mechanism reported in Fig. 1 does not point toward this residue and is relatively far from it (>4 Å for all binding poses). To explain SB-3CT mechanism of inhibition,18 the authors faced the same problem and undertook MD simulations to explore the conformational surface of MMP-2/SB-3CT complex and to select a conformation in which the deprotonation of SB-3CT by Glu404 was feasible.
We have performed a comparative MD study of 2f and SB-3CT complexed to MMP-2 with the aim of exploring the ability of these inhibitors to adopt conformations, within the MMP-2 catalytic cleft, that could allow their deprotonation by Glu404 and the subsequent opening of the thiirane ring (see below).
• Docking of thiolate derivatives 7a–f.
(E) and (Z) stereoisomers of 1 and 7a–f, resulting from the opening of respectively SB-3CT and 2a–f, were docked using the same MMP-2 structure coming from previous MD simulations.28 Both diastereoisomers of all compounds gave similar binding to MMP-2 with close and excellent binding scores. The most energetically favoured poses of those derivatives (see Fig. 5 for the two isomers of 7f) showed the expected interactions: a sulfone oxygen establishes H-bonds to Leu191 and Ala192 backbones, the P1′ fragment goes deep into S1′ pocket establishing various VdW contacts and the thiolate anion coordinates the catalytic Zn2+, forming a pseudo trigonal-based bipyramid, from which a ligand (water) is missing.
 | ||
| Fig. 5 Most energetically favoured binding modes for (E)-7f (A) and (Z)-7f (B) in MMP-2. H-bonds (orange dots) and alkene hydrogens are depicted. Zn2+–sulphur distances are 2.20 Å (A) and 2.19 Å (B), respectively. | ||
These results suggest that the opening of 2a–f would lead to potent MMP-2 inhibitors.
Molecular dynamics simulations
With the aim of rationalizing the difference of inhibitory activity experimentally observed between 2f and SB-3CT, we run MD simulations for these two inhibitors. As it has been reported in the literature, the inhibitory capacity of this type of compounds comes from their ability to react with Glu404. This is only possible in an initial phase of the binding, in which the thiirane sulphur atom is not coordinating the catalytic Zn ion.18,19,36 Therefore 20 ns MD simulations were carried out in order to assess the dynamic stability of the binding mode of the R-enantiomer of SB-3CT in complex with MMP-2 and compare it to (R)-2f (Fig. 6A and B, respectively). We chose to study only the R enantiomer of both compounds as both enantiomers of SB-3CT showed similar inhibitory activity,33 and because more information about MD studies with (R)-SB-3CT can be found in the literature.18 The main stability parameter we focused on was the H-bond established between the sulfone moiety of the two ligands and the backbone NH of both Leu191 and Ala192. Thus, we measured the hydrogen bonding distances between the backbone NH of both residues and the nearest sulfone oxygen atom (black and grey line in Fig. 6). While in the complex of MMP-2 with (R)-SB-3CT the distance and the bound conformation remain very stable during the whole simulation, in the complex with compound (R)-2f the interaction distances fluctuate and the interaction is lost at the end of the simulation time (Fig. S1†). In addition, we measured the distance between the carboxylic group of Glu404 and the hydrogen that has to be abstracted in (R)-SB-3CT and (R)-2f in order to form thiolates 1 and 7f, respectively. We found that the distance fluctuated considerably for (R)-2f even during the first 16 ns of simulation (in which the interactions with Leu191 and Ala192 were maintained), while remained much more stable for (R)-SB-3CT. As a consequence, the probability for the proton abstraction and further ring opening reaction must be smaller for (R)-2f than for (R)-SB-3CT. | ||
| Fig. 6 Time evolution of the interaction distances measured (Å) in the complexes of MMP2 with (A) (R)-SB-3CT and (B) (R)-2f between the sulfone moiety and Leu191 and Ala192 (black and grey lines respectively), the reactive hydrogen and Glu404 (blue line) and the sulphur atom of the thiirane ring and the catalytic zinc ion (red line). | ||
The distance between the sulphur atom of the thiirane and the catalytic zinc ion also fluctuates much more in the MMP-2/(R)-2f complex.
This different behaviour arises from the smaller size and the flexibility of (R)-SB-3CT compared to compound (R)-2f. The four aromatic rings present on (R)-2f confer a rigid planar conformation to the molecule, which does not fit in the S1′ pocket as good as the (R)-SB-3CT conformation. As a consequence, the sulfone moiety of compound (R)-2f, although establishing apparently stable interactions with the backbone NH of Leu191 and Ala192, protrudes a little more out of the binding pocket than (R)-SB-3CT (Fig. S2†), inducing the observed fluctuation of the distance to Glu404. This increased mobility of the thiirane end of compound (R)-2f, that goes along with a greater distance fluctuation between the hydrogen and the carbonyl of Glu404, reduces the probability of a nucleophilic attack on the hydrogen compared to that of compound (R)-SB-3CT and, therefore, also reduces the reaction rate of the thiirane opening; all of which is in line with the experimental findings.
From these results, the P1′ chain appears critical in the potency of the inhibitors. P1′ fragments that gave us excellent results with the hydroxamate ZBG (such as in compounds 2a and 2d)27,28 are inadequate when using the thiirane latent ZBG. Insufficient interactions at the S1′ pocket – for instance in azide 6 that misses a P1′ fragment – or too rigid/bulky P1′ groups may destabilize the inhibitor binding and thus affect the turn over of the thiirane opening, as suggested by our comparative MD simulation studies. This hypothesis could explain the weak potency of inhibitors 8 (previously reported25) and inhibitors 2a and 2b that bear bulky P1′ fragments.
Conclusions
We synthesized a series of clicked thiiranes as new gelatinases inhibitors. Our hit (2f) displayed submicromolar IC50 against MMP-2 in a colorimetric assay, strongly improving the potency of other clicked thiirane derivatives previously reported. To understand the difference of potency between 2f and the reference thiirane inhibitor SB-3CT, we carried out MD simulations for both inhibitors complexed to the MMP-2 catalytic domain. We propose that the longer and more rigid P1′ fragment present in 2f is responsible for the lower stability of this compound in MMP-2, causing a reduction on the reaction rate of the thiirane opening. These results put forward the relevance of the P1′ fragment in the potency of this type of inhibitors. We are now combining computational simulations with our click approach to better understand the effect of the P1′ fragment on the reaction rate of the thiirane opening within the MMP, and with the final aim of developing clicked thiirane derivatives with improved potency.Experimental section
Chemistry
4-(Allylthio)aniline (3). To a suspension of sodium hydride (1.53 g, 38.3 mmol) in dry DMF (15 mL) at 0 °C, was slowly added a solution of p-aminothiophenol (4.0 g, 32.0 mmol) in dry DMF. The resulting dark mixture was stirred 10 minutes at 0 °C under argon and a solution of allyl chloride (2.7 g, 35.1 mmol) in dry DMF was added dropwise. The reaction mixture was then allowed to reach RT and stirred overnight under argon. The reaction was concentrated and the residue was partitioned between water and EtOAc. The organic layer was washed with water (×3) and brine, dried over MgSO4 (anhydrous), filtered and evaporated to afford 5.1 g of brown oil. The crude product was purified on silica gel (9
:1 to 7:3 hexane–EtOAc) to afford N-alkylated by-product (860 mg, 16%) and 3 (3.6 g, 68%) as a pale orange oil. 1H NMR (300 MHz, CDCl3) δ 3.38 (dt, J = 7.1, 1.1, 2H), 3.70 (br s, 2H), 4.92–5.00 (m, 2H), 5.77–5.90 (m, 1H), 6.60 (“d”, J = 8.6 Hz, 2H), 7.23 (“d”, J = 8.6 Hz, 2H); 13C NMR (75.4 MHz, CDCl3) δ 40.0 (CH2 in DEPT-135), 115.5 (CH), 117.1 (CH2), 122.8, 134.4 (CH), 134.6 (CH), 146.2; MS (ESI) bad ionization.
Allyl(4-azidophenyl)sulfane (4). To a solution of 3 (3.5 g, 21.2 mmol) in dry MeCN (30 mL) was added tert-butyl nitrite (2.4 g, 23.3 mmol) at 0 °C under argon. The mixture was stirred 10 min at 0 °C and TMSN3 (2.7 g, 23.3 mmol) was slowly added. After stirring the mixture for 30 min at 0 °C and 2 h at RT, the reaction mixture was concentrated and the residue was partitioned between water and EtOAc. The organic layer was washed with water and brine, dried over MgSO4 (anhydrous), filtered and evaporated to afford 3.8 g of brown oil. The crude product was filtered through silica gel, eluting with 10% CH3Cl in hexane, to afford 4 (3.4 g, 84%) as a pale orange oil. IR (KBr): 2159 (N3 stretch) cm−1; 1H NMR (300 MHz, CDCl3) δ 3.50 (dt, J = 7.2 1.1 Hz, 2H), 5.04–5.10 (m, 2H), 5.78–5.92 (m, 1H), 6.96 (“d”, J = 8.7, 2H), 7.35 (“d”, J = 8.7, 2H); 13C NMR (75.4 MHz, CDCl3) δ 38.2, 117.9, 119.6, 132.0, 132.4, 133.6, 138.8; anal. calcd for C9H9N3S: C, 56.52; H, 4.74; N, 21.97; S, 16.77. Found: C, 55.54; H, 5.30; N, 21.03; S, 16.68%.
2-(((4-Azidophenyl)sulfonyl)methyl)oxirane (5). To a solution of 4 (3.0 g, 15.7 mmol) in CH2Cl2 (150 mL) was added mCPBA (16.2 g, 94.1 mmol, 6.00 eq.) at 0 °C under argon. The mixture was stirred at RT under argon. After 2 days, TLC showed a mixture of starting material, m-chlorobenzoic acid (mCBA) and two intermediates. A sample was taken in CH2Cl2 and washed with 2% NaOH solution (×3), water and brine. After drying (MgSO4 anhydrous) and evaporating the organic layer, 1H NMR showed mCBA, starting material, ∼10% of epoxide compound(s), and a mixture of allyl derivatives. To remove mCBA, the reaction mixture was worked-up as above and plugged on silica pad (eluting with CH2Cl2) and dried. The yellow oil obtained was set back to react using the same conditions described above. TLC showed progress. After 2 days this manipulation was repeated. After 3 more days, the reaction was stopped and worked-up as previously described to give 2.3 g of yellow oil. The crude product was purified on silica gel (9
:1 to 7:3 hexane–EtOAc) to afford 5 (1.4 g, 37%) as a yellow oil. IR (KBr): 2140 (N3 stretch) cm−1; 1H NMR (300 MHz, CDCl3) δ 2.45 (dd, J = 4.8, 2.0 Hz, 1H), 2.79–2.82 (m, 1H), 3.24–3.37 (m, 3H), 7.20 (d, J = 8.8 Hz, 2H), 7.93 (d, J = 8.8 Hz, 2H); 13C NMR (75.4 MHz, CDCl3) δ 46.0, 59.8, 119.8, 130.4, 135.3, 146.5.
2-(((4-Azidophenyl)sulfonyl)methyl)thiirane (6). A solution of 5 (1.3 g, 5.4 mmol) and thiourea (827 mg, 10.9 mmol) in 1
:1 MeOH–CH2Cl2 (30 mL) was stirred overnight at RT under argon. The reaction mixture was concentrated and the residue was partitioned between water and EtOAc. The organic layer was washed with water, dried over MgSO4 (anhydrous), filtered and evaporated to afford 1.2 g of a white solid. The crude product was purified on silica gel, eluting with 8:2 hexane–EtOAc to afford 6 (1.1 g, 81%) as a white solid. IR (KBr): 2119 (N3 stretch) cm−1; 1H NMR (300 MHz, CDCl3) δ 2.13 (dd, J = 5.1 1.7 Hz, 1H), 2.53 (dd, J = 6.1 1.5 Hz, 1H), 3.02–3.10 (m, 1H), 3.23 (dd, J = 14.3 7.4 Hz, 1H), 3.48 (dd, J = 14.3 5.9 Hz, 1H), 7.20 (d, J = 8.7 Hz, 2H), 7.91 (d, J = 8.7 Hz, 2H); 13C NMR (75.4 MHz, CDCl3) δ 24.2 (CH2 in DEPT-135), 26.1 (CH), 62.7 (CH2), 119.8 (CH), 130.6 (CH), 134.8, 146.6.
N,N-Dimethyl-4-(1-(4-((thiiran-2-ylmethyl)sulfonyl)phenyl)-1H-1,2,3-triazole-4-yl)aniline (2a). A mixture of 6 (150 mg, 0.59 mmol), 4-ethynyl-N,N-dimethylaniline (102 mg, 0.70 mmol), freshly made aqueous 0.1 M CuSO4·5H2O solution (350 μL, 0.03 mmol, 0.06 eq.) and 0.5 M sodium ascorbate solution (234 μL, 0.12 mmol, 0.20 eq.) in 1
:1 H2O–tBuOH (3 mL) was stirred at RT under argon for 24 h. The reaction mixture was partitioned between EtOAc and water. A brown precipitate appeared in the organic layer. The aqueous part was discarded and the solvent was evaporated. The resulting brown solid was triturated in Et2O and recrystallized from EtOAc to afford 2a (53 mg, 22%) as an orange solid, mp 226–227 °C. 1H NMR (300 MHz, CDCl3) δ 2.23 (dd, J = 5.3, 1.0 Hz, 1H), 2.58 (dd, J = 5.3, 1.0 Hz, 1H), 2.97 (s, 6H), 3.02–3.11 (m, 1H), 3.71–3.85 (m, 2H), 6.84 (d, J = 8.9 Hz, 2H), 7.77 (d, J = 8.9 Hz, 2H), 8.16 (d, J = 8.9 Hz, 2H), 8.27 (d, J = 8.9 Hz, 2H), 9.30 (s, 1H); 13C NMR (75.4 MHz, CDCl3) δ 23.9, 26.8, 39.9, 60.4, 112.3, 117.4, 117.6, 120.0, 126.3, 130.3, 137.8, 140.4, 148.4, 150.4; MS (ESI) 401.0 [M + H]+; anal. calcd for C19H20N4O2S2: C, 56.98; H, 5.03; N, 13.99; S, 16.01. Found: C, 57.03; H, 5.07; N, 13.91; S, 15.72%.
4-(4-Pentylphenyl)-1-(4-((thiiran-2-ylmethyl)sulfonyl)phenyl)-1H-1,2,3-triazole (2b). A mixture of 6 (100 mg, 0.39 mmol), 1-ethynyl-4-pentylbenzene (81 mg, 0.470 mmol), freshly made aqueous 0.1 M CuSO4·5H2O solution (235 μL, 0.02 mmol, 0.06 eq.) and 0.5 M sodium ascorbate solution (157 μL, 0.08 mmol, 0.20 eq.) in MeCN (2 mL) was stirred at RT under argon for 24 h. Water was added and the precipitate was filtered, washed with water and CH2Cl2 to give 104 mg of a yellow solid. The crude product was consequently purified by column chromatography on silica gel (1–5% MeOH–CH2Cl2) to give 2b (25 mg, 15%) as a white solid, mp 187–188 °C. 1H NMR (300 MHz, CDCl3) δ 0.90 (“t”, 3H), 1.32–1.37 (m, 4H), 1.61–1.71 (m, 2H), 2.18 (dd, J = 5.1 1.8 Hz, 1H), 2.57 (dd, J = 6.2 1.8 Hz, 1H), 2.66 (t, J = 7.9 Hz, 2H), 3.07–3.15 (m, 1H), 3.33 (dd, J = 14.4 7.2 Hz; 1H), 3.52 (dd, J = 14.4 6.2 Hz, 1H), 7.30 (d, J = 8.2 Hz, 2H), 7.83 (d, J = 8.2 Hz, 2H), 8.08 (d, J = 8.9 Hz, 2H), 8.14 (d, J = 8.9 Hz, 2H), 8.27 (s, 1H); 13C NMR (75.4 MHz, CDCl3) δ 14.1, 22.7, 24.1, 26.0, 31.2, 31.6, 35.9, 62.7, 116.9, 120.0, 120.7, 126.1, 129.2, 130.7, 138.5, 141.2, 144.2, 149.5; MS (ESI) 428.0 [M + H]+. Purity by HPLC-MS: 87%.
4-([1,1′-Biphenyl]-4-yl)-1-(4-((thiiran-2-ylmethyl)sulfonyl)phenyl)-1H-1,2,3-triazole (2c). Following the general procedure, starting from 4-ethynyl-1,1′-biphenyl, triazole 2c (70 mg, 41%) was obtained as a beige solid, mp 236–237 °C (from MeCN). 1H NMR (300 MHz, CDCl3) δ 2.23 (d, J = 4.8 Hz, 1H), 2.58 (d, J = 5.3 Hz, 1H), 3.03–3.11 (m, 1H), 3.72–3.86 (m, 2H), 7.40 (t, J = 7.3 Hz, 1H), 7.50 (t, J = 7.3 Hz, 2H), 7.76 (d, J = 7.3 Hz, 2H), 7.85 (d, J = 8.4 Hz, 2H), 8.06 (d, J = 8.4 Hz, 2H), 8.20 (d, J = 8.8 Hz, 2H), 8.31 (d, J = 8.8 Hz, 2H), 9.58 (s, 1H); 13C NMR (75.4 MHz, CDCl3) δ 23.9, 26.8, 60.4, 120.0, 120.3, 126.0, 126.6, 127.3, 127.7, 128.9, 129.0, 130.3, 138.2, 139.4, 140.1, 140.3, 147.4; MS (ESI) 434.1 [M + H]+. Anal. calcd for C23H19N3O2S2: C, 63.72; H, 4.41; N, 9.69; S, 14.79. Found: C, 63.64; H, 4.50; N, 9.80; S, 13.79%.
4-(4-Methoxyphenyl)-1-(4-((thiiran-2-ylmethyl)sulfonyl)phenyl)-1H-1,2,3-triazole (2d). Following the general procedure, starting from 1-ethynyl-4-methoxybenzene, triazole 2d (55 mg, 36%) was obtained as a white solid, mp 207–208 °C. 1H NMR (300 MHz, CDCl3) δ 2.23 (d, J = 4.5 Hz, 1H), 2.57 (d, J = 5.6 Hz, 1H), 3.02–3.10 (m, 1H), 3.71–3.81 (m, 2H), 3.82 (s, 3H), 7.09 (d, J = 8.8 Hz, 2H), 7.89 (d, J = 8.8 Hz, 2H), 8.17 (d, J = 8.8 Hz, 2H), 8.28 (d, J = 8.8 Hz, 2H), 9.40 (s, 1H); 13C NMR (75.4 MHz, CDCl3) δ 23.9, 26.8, 55.2, 60.4, 114.5, 118.8, 120.2, 122.3, 126.8, 130.3, 138.1, 140.3, 147.7, 159.5; MS (ESI) 388.1 [M + H]+. Purity by HPLC-MS: 89%.
4-(3-Fluorophenyl)-1-(4-((thiiran-2-ylmethyl)sulfonyl)phenyl)-1H-1,2,3-triazole (2e). Following the general procedure, starting from 1-ethynyl-3-fluorobenzene, triazole 2e (51 mg, 34%) was obtained as a beige solid, mp 189–190 °C. 1H NMR (300 MHz, DMSO-d6) δ 2.23 (d, J = 5.3 Hz, 1H), 2.58 (d, J = 5.3 Hz, 1H), 3.05–3.09 (m, 1H), 3.72–3.86 (m, 2H), 7.26 (td, J = 8.9, 2.4 Hz, 1H), 7.55–7.62 (m, 1H), 7.76 (d, J = 10.1 Hz, 1H), 7.82 (d, J = 7.7 Hz, 1H), 8.20 (d, J = 8.8 Hz, 2H), 8.27 (d, J = 8.8 Hz, 2H), 9.59 (s, 1H); 13C NMR (75.4 MHz, DMSO-d6) δ 23.9, 26.8, 60.4, 112.0 (d, 2JCF = 23.0 Hz), 115.2 (d, 2JCF = 21.2 Hz), 120.3, 120.7, 121.5, 130.4, 131.3 (d, 3JCF = 8.7 Hz), 132.2 (d, 3JCF = 8.6 Hz), 138.3, 140.2, 146.6, 162.6 (d, 1JCF = 243 Hz); MS (ESI) 375.9 [M + H]+. Purity by HPLC-MS: 86%.
4-(4-(1-(4-((Thiiran-2-ylmethyl)sulfonyl)phenyl)-1H-1,2,3-triazole-4-yl)phenyl)pyridine (2f). Following the general procedure, starting from 4-(4-ethynylphenyl)pyridine,28 triazole 2f (42 mg, 25%) was obtained as a white solid, mp 213–214 °C (from MeCN). 1H NMR (300 MHz, DMSO-d6) δ 2.24 (d, J = 5.2 Hz, 1H), 2.58 (d, J = 5.7 Hz, 1H), 3.03–3.1 (m, 1H), 3.72–3.87 (m, 2H), 7.83 (br s, 2H), 7.99 (d, J = 8.3 Hz, 2H), 8.12 (d, J = 8.3 Hz, 2H), 8.20 (d, J = 8.7 Hz, 2H), 8.30 (d, J = 8.7 Hz, 2H), 8.76 (br s, 1–2H), 9.62 (s, 1H); 13C NMR (75.4 MHz, DMSO-d6) δ 22.9, 25.8, 59.4, 119.3, 119.4, 120.0, 125.1, 126.5, 129.4, 129.7, 135.9, 137.3, 139.3, 145.2, 146.1, 149.3; MS (ESI) 435.0 [M + H]+. Anal. calcd for C22H18N4O2S2: C, 60.81; H, 4.18; N, 12.89; S, 14.76. Found: C, 60.55; H, 4.24; N, 12.84; S, 14.01%.
MMPs inhibition assays
MMPs activity measurements were performed using MMP Inhibitor Profiling Kit purchased from Enzo Life Science International, Inc., and following the manufacturer's protocol with slight modifications. Proteolytic activity was measured using a thiopeptide substrate (Ac-PLG-[2-mercapto-4-methylpentanoyl]-LG-OC2H5) where the MMP cleavage site peptide bond has been replaced by a thioester bond.37,38 Hydrolysis of this bond by MMP produces a sulfhydryl group that reacts with DTNB to form 2-nitro-5-thiobenzoic acid, which was detected by its absorbance at 414 nm (microplate photometer Thermo Scientific Multiscan FC). Enzyme reactions were carried out at 37 °C in a 100 μL final volume of solutions, where the catalytic domains of the corresponding MMP were incubated 45 min or 2 h in triplicate with at least seven concentrations of inhibitors. The compounds were dissolved in DMSO, with 2% final concentration in each well. The negative control (enzyme + substrate contained the same amount of DMSO solvent). The assay buffer contained the following components: 50 mM HEPES, 10 mM CaCl2, 0.05% Brij-35 and 1 mM DTNB at pH 7.5. After addition of substrate, the increase of absorbance was recorded in 1 min time intervals for 30 min. Data were plotted as OD versus time for each sample, in order to obtain the reaction velocity (V) in OD per min. The percentage of residual activity for each compound was calculated using the following formula: % of remaining activity = (V in the presence of inhibitor/Vcontrol) × 100. An inhibitor, NNGH, was included as a prototypic control inhibitor.39 The concentration of compound that provided 50% inhibition of enzymatic activity (IC50) was determined by semi-logarithmic dose–response plots (Graph Pad Prism 5.0 for Windows, Graph Pad Software Inc., San Diego, California, USA, 2007).NMR studies
All spectra were recorded at 300 K with a Bruker Avance 600 MHz spectrometer equipped with a 5 mm TCI cryoprobe. A typical NMR sample contained a concentration of 5 μM of MMP-2 and 100 μM of ligand (from a 50 mM stock in DMSO-d6), in an approximate protein–ligand ratio of 1:20, optimal for the waterLOGSY experiments. To 450 μL of sample (buffer: 10 mM deuterated-Tris/HCl pH 7.4 with 50 mM NaCl, 0.02% NaN3, 100 μM CaCl2 and 100 μM ZnCl2) 25 μL of D2O were added for locking purposes, and 15 μL of DMSO-d6 to increase the solubility of the compounds. BiPS (2R-[(4-biphenylsulfonyl)amino]-N-hydroxy-3-phenylpropinamide; VWR, Barcelona, Spain) was used for the competition experiments from a 50 mM stock in DMSO-d6. For each sample, 1D 1H and waterLOGSY (direct interaction and competition) experiments were recorded. 8k points were used for a sweep width of 9600 Hz and a total of 512 scans were accumulated for the waterLOGSY experiment. In these experiments, the large bulk water magnetization is partially transferred via the protein–ligand complex to the free ligand in a selective manner. A non-interacting compound results in negative resonances, whereas protein–ligand interactions are characterized by positive signals or by a reduction in the negative signals obtained in the absence of the protein.
Computational methods
To relax and ensure a correct opening of S1′ in 1CK7, we used a structure extracted from a previously reported MD simulation of a MMP-2 hydroxamate inhibitor bearing the same P1′ fragment as 2a.28
Acknowledgements
This work was supported by the Spanish Ministry of Science and Innovation (SAF2008-00945, CTQ2011-24741, SAF2011-28350). Grants to B. F. and K. F. from Fundación Universitaria San Pablo CEU are also acknowledged. We thank Synthelia Organics SL. for HPLC-MS experiments.Notes and references
- H. Nagase and J. F. Woessner, J. Biol. Chem., 1999, 274, 21491–21494 CrossRef CAS PubMed.
- C. E. Brinckerhoff and L. M. Matrisian, Nat. Rev. Mol. Cell Biol., 2002, 3, 207–214 CrossRef CAS PubMed.
- D. Rodríguez, C. J. Morrison and C. M. Overall, BBA, Biochim. Biophys. Acta, Mol. Cell Res. Mol. Cell Res., 2010, 1803, 39–54 CrossRef PubMed.
- T. Klein and R. Bischoff, Amino Acids, 2011, 41, 271–290 CrossRef CAS PubMed.
- M. D. Martin and L. M. Matrisian, Cancer Metastasis Rev., 2007, 26, 717–724 CrossRef CAS PubMed.
- L. M. Coussens, B. Fingleton and L. M. Matrisian, Science, 2002, 295, 2387–2392 CrossRef CAS PubMed.
- B. Fingleton, Semin. Cell Dev. Biol., 2008, 19, 61–68 CrossRef CAS PubMed.
- K. Maskos, Biochimie, 2005, 87, 249–263 CrossRef CAS PubMed.
- L. Devel, B. Czarny, F. Beau, D. Georgiadis, E. Stura and V. Dive, Biochimie, 2010, 92, 1501–1508 CrossRef CAS PubMed.
- J. A. Jacobsen, J. L. Major Jourden, M. T. Miller and S. M. Cohen, BBA, Biochim. Biophys. Acta, Mol. Cell Res. Mol. Cell Res., 2010, 1803, 72–94 CAS.
- P. Serra, M. Bruczko, J. Zapico, A. Puckowska, M. García, S. Martín-Santamaría, A. Ramos and B. de Pascual-Teresa, Curr. Med. Chem., 2012, 19, 1036–1064 CrossRef CAS.
- R. Yadav, S. Gupta, P. Sharma and V. Patil, Curr. Med. Chem., 2011, 18, 1704–1722 CrossRef CAS.
- S. Brown, M. M. Bernardo, Z. H. Li, L. P. Kotra, Y. Tanaka, R. Fridman and S. Mobashery, J. Am. Chem. Soc., 2000, 122, 6799–6800 CrossRef CAS.
- A. Krüger, M. J. E. Arlt, M. Gerg, C. Kopitz, M. M. Bernardo, M. Chang, S. Mobashery and R. Fridman, Cancer Res., 2005, 65, 3523–3526 CrossRef PubMed.
- R. D. Bonfil, A. Sabbota, S. Nabha, M. M. Bernardo, Z. Dong, H. Meng, H. Yamamoto, S. R. Chinni, I. T. Lim, M. Chang, S. Mobashery and R. Fridman, Int. J. Cancer, 2006, 118, 2721–2726 CrossRef CAS PubMed.
- O. Kleifeld, L. P. Kotra, D. C. Gervasi, S. Brown, M. M. Bernardo, R. Fridman, S. Mobashery and I. Sagi, J. Biol. Chem., 2001, 276, 17125–17131 CrossRef CAS PubMed.
- C. Forbes, Q. Shi, J. F. Fisher, M. Lee, D. Hesek, L. I. Llarrull, M. Toth, M. Gossing, R. Fridman and S. Mobashery, Chem. Biol. Drug Des., 2009, 74, 527–534 CAS.
- P. Tao, J. F. Fisher, Q. Shi, T. Vreven, S. Mobashery and H. B. Schlegel, Biochemistry, 2009, 48, 9839–9847 CrossRef CAS PubMed.
- P. Tao, J. F. Fisher, Q. Shi, S. Mobashery and H. B. Schlegel, J. Phys. Chem. B, 2009, 114, 1030–1037 CrossRef PubMed.
- M. Lee, A. Villegas-Estrada, G. Celenza, B. Boggess, M. Toth, G. Kreitinger, C. Forbes, R. Fridman, S. Mobashery and M. Chang, Chem. Biol. Drug Des., 2007, 70, 371–382 CAS.
- G. Celenza, A. Villegas-Estrada, M. Lee, B. Boggess, C. Forbes, W. R. Wolter, M. A. Suckow, S. Mobashery and M. Chang, Chem. Biol. Drug Des., 2008, 71, 187–196 CAS.
- M. Gooyit, M. Lee, D. Hesek, B. Boggess, A. G. Oliver, R. Fridman, S. Mobashery and M. Chang, Chem. Biol. Drug Des., 2009, 74, 535–546 CAS.
- S. A. Testero, M. Lee, R. T. Staran, M. Espahbodi, L. I. Llarrull, M. Toth, S. Mobashery and M. Chang, ACS Med. Chem. Lett., 2011, 2, 177–181 CrossRef CAS.
- M. Gooyit, M. Lee, V. A. Schroeder, M. Ikejiri, M. A. Suckow, S. Mobashery and M. Chang, J. Med. Chem., 2011, 54, 6676–6690 CrossRef CAS PubMed.
- S. A. Testero, L. I. Llarrull, J. F. Fisher, M. Chang, S. Mobashery and S. Giordano, ARKIVOC, 2011, 221–236 CrossRef CAS.
- E. Morgunova, A. Tuuttila, U. Bergmann, M. Isupov, Y. Lindqvist, G. Schneider and K. Tryggvason, Science, 1999, 284, 1667–1670 CrossRef CAS.
- J. M. Zapico, P. Serra, J. Garcia-Sanmartin, K. Filipiak, R. J. Carbajo, A. K. Schott, A. Pineda-Lucena, A. Martinez, S. Martin-Santamaria, A. Ramos and B. de Pascual-Teresa, Org. Biomol. Chem., 2011, 9, 4587–4599 CAS.
- B. Fabre, K. Filipiak, J. M. Zapico, N. Díaz, R. J. Carbajo, A. K. Schott, M. P. Martínez-Alcázar, D. Suárez, A. Pineda-Lucena, A. Ramos and B. de Pascual-Teresa, Org. Biomol. Chem., 2013, 11, 6623–6641 CAS.
- I. Schechter and A. Berger, Biochem. Biophys. Res. Commun., 1967, 27, 157 CrossRef CAS.
- B. Fabre, K. Filipiak, N. Díaz, J. M. Zapico, D. Suárez, A. Ramos and B. de Pascual-Teresa, ChemBioChem, 2014, 15, 399–412 CrossRef CAS PubMed.
- C. M. Overall and O. Kleifeld, Nat. Rev. Cancer, 2006, 6, 227–239 CrossRef CAS PubMed.
- M. M. Bernardo, S. Brown, Z. H. Li, R. Fridman and S. Mobashery, J. Biol. Chem., 2002, 277, 11201–11207 CrossRef CAS PubMed.
- M. Lee, M. M. Bernardo, S. O. Meroueh, S. Brown, R. Fridman and S. Mobashery, Org. Lett., 2005, 7, 4463–4465 CrossRef CAS PubMed.
- P. C. Chen, R. E. Wharton, P. A. Patel and A. K. Oyelere, Bioorg. Med. Chem., 2007, 15, 7288–7300 CrossRef CAS PubMed.
- C. Dalvit, G. Fogliatto, A. Stewart, M. Veronesi and B. Stockman, J. Biomol. NMR, 2001, 21, 349–359 CrossRef CAS.
- J. Zhou, P. Tao, J. F. Fisher, Q. Shi, S. Mobashery and H. B. Schlegel, J. Chem. Theory Comput., 2010, 6, 3580–3587 CrossRef CAS PubMed.
- H. Weingarten, R. Martin and J. Feder, Biochemistry, 1985, 24, 6730–6734 CrossRef CAS.
- H. Weingarten and J. Feder, Anal. Biochem., 1985, 147, 437–440 CrossRef CAS.
- L. J. MacPherson, E. K. Bayburt, M. P. Capparelli, B. J. Carroll, R. Goldstein, M. R. Justice, L. Zhu, S. Hu, R. A. Melton, L. Fryer, R. L. Goldberg, J. R. Doughty, S. Spirito, V. Blancuzzi, D. Wilson, E. M. O'Byrne, V. Ganu and D. T. Parker, J. Med. Chem., 1997, 40, 2525–2532 CrossRef CAS PubMed.
- F. Molina, L. L. Johnson, D. F. Ortwine, A. Pavlovsky, J. R. Rubin, R. W. Skeean, A. D. White, C. Humblet, D. J. Hupe and T. L. Blundell, Croat. Chem. Acta, 1999, 72, 575–591 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, T. Daniel, M. P. Repasky, E. H. Knoll, M. Shelley and J. K. Perry, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- Schrödinger Suite 2011 Protein Preparation Wizard, Epik version 2.2, Schrödinger, LLC, New York, NY, 2011 Search PubMed; Impact version 5.7, Schrödinger, LLC, New York, NY, 2011 Search PubMed; Prime version 3.0, Schrödinger, LLC, New York, NY, 2011 Search PubMed.
- LigPrep, version 2.5, Schrödinger, LLC, New York, NY, 2011 Search PubMed.
- J. C. Gordon, J. B. Myers, T. Folta, V. Shoja, L. S. Heath and A. Onufriev, Nucleic Acids Res., 2005, 33, W368–W371 CrossRef CAS PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, J. Montgomery Jr, T. Vreven, K. Kudin and J. Burant, Gaussian, Inc., Wallingford, CT, 2004.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS PubMed.
- J. Aaqvist, J. Phys. Chem., 1990, 94, 8021–8024 CrossRef CAS.
- AMBER 10, University of California, San Francisco.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS PubMed.
- M. A. Lill, Biochemistry, 2011, 50, 6157–6169 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of the compounds, competition waterLOGSY experiment for 2e, the method used to obtain the 1CK7 structure with relaxed S1′ pocket, as well as the starting and final structures of the MD simulations are found in ESI. See DOI: 10.1039/c3ra46402d |
| This journal is © The Royal Society of Chemistry 2014 |