
A chiral macrocyclic receptor for sulfate anions with CD signals†
Abstract
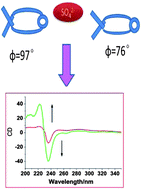
A novel macrocyclic binaphthalene derivative (L) containing amide and triazole units has been synthesized by “click” reaction. The binding behaviors of this receptor toward anions have been studied by 1H NMR titration, circular dichroism (CD) spectroscopy. The tetrahedral sulfate anion predominantly interacts with L through hydrogen bonds which could tune the dihedral angle between the two naphthalene rings and supply tunable CD output signals to form chiral receptor.
 Please wait while we load your content...
Please wait while we load your content...

